
-
摘要: 目的 分析GSDME基因变异耳聋家系的遗传、听力学特点, 探索其发病特点和致病机制, 以期为患者提供遗传咨询及干预指导。方法 纳入来自中国聋病基因组计划项目的6个迟发性非综合征型听力损失家系, 通过纯音测听、声导抗、言语识别率、听性脑干反应和畸变产物耳声发射等听力学检测评估患者听力水平, 结合病史采集及体格检查分析先证者及其家系成员间的表型差异。应用二代测序检测先证者致病基因, 并使用Sanger测序对家系其他成员进行变异位点验证, 依据美国遗传学与基因组医学委员会指南进行致病性分析。同时, 结合国内外GSDME研究进展, 探讨可能的致聋机制。结果 在6个迟发性非综合征型听力损失家系中, 共30例有听力损失表型, 发病年龄10~50岁(27.88±9.74岁)。遗传学分析鉴定4个GSDME基因剪切变异, 其中2个变异为新发现的变异, 分别是c.991-7C>G和c.1183+1G>T, 且c.991-7C>G是GSDME新发变异。另外2个变异为已报道的GSDME剪切变异, 分别是c.991-1G>C和c.991-15_991-13del, 且c.991-15_991-13del在3个家系中检出。基因型-表型相关分析发现携带c.991-7C>G和c.1183+1G>T变异的先证者均表现为高频下降为主的听力损失表型, 相同变异的3个家系的先证者听力损失程度不一且听力损失的年下降率高于既往报道的0.94 dB HL/年。此外, 随访发现6个家系内的先证者, 有4例接受干预(66.67%), 但干预效果不一。结论 本研究分析GSDME变异相关的6个迟发性非综合征型听力损失家系, 共鉴定4个剪切变异, 其中1个为国内外首个GSDME新发变异, 听力学分析发现患者多在10岁后出现渐进性听力损失, 且不同干预的效果存在差异。
-
关键词:
- 常染色体显性耳聋5型 /
- GSDME基因 /
- 遗传咨询 /
- 干预
Abstract: Objective To dentify the genetic and audiological characteristics of families affected by late-onset hearing loss due to GSDMEgene mutations, aiming to explore clinical characteristics and pathogenic mechanisms for providing genetic counseling and intervention guidance.Methods Six families with late-onset hearing loss from the Chinese Deafness Genome Project were included. Audiological tests, including pure-tone audiometry, acoustic immittance, speech recognition scores, auditory brainstem response, and distortion product otoacoustic emission, were applied to evaluate the hearing levels of patients. Combining with medical history and physical examination to analyze the phenotypic differences between the probands and their family members. Next-generation sequencing was used to identify pathogenic genes in probands, and validations were performed on their relatives by Sanger sequencing. Pathogenicity analysis was performed according to the American College of Medical Genetics and Genomics Guidelines. Meanwhile, the pathogenic mechanisms of GSDME-related hearing loss were explored combining with domestic and international research progress.Results Among the six families with late-onset hearing loss, a total of 30 individuals performed hearing loss. The onset of hearing loss in these families ranged from 10 to 50 years(mean age: 27.88±9.74 years). In the study, four splicing mutations of the GSDME were identified, including two novel variants: c. 991-7C>G and c. 1183+1G>T. Significantly, the c. 991-7C>G was a de novo variant. The others were previously reported variants: c. 991-1G>C and c. 991-15_991-13del, the latter was identified in three families. Genotype-phenotype correlation analysis revealed that probands with the c. 991-7C>G and c. 1183+1G>T performed a predominantly high-frequency hearing loss. The three families carrying the same mutation exhibited varying degrees of hearing loss, with an annual rate of hearing deterioration exceeding 0.94 dB HL/year. Furthermore, follow-up of interventions showed that four of six probands received intervention(66.67%), but the results of intervention varied.Conclusion The study analyzed six families with late-onset non-syndromic hearing loss linked to GSDME mutations, identifying four splicing variants. Notably, c. 991-7C>G is the first reported de novo variant of GSDME globally. Audiological analysis revealed that the age of onset generally exceeded 10 years, with variable effectiveness of interventions.-
Key words:
- Autosomal dominant deafness 5 /
- GSDME /
- Genetic counseling /
- Intervention
-
-
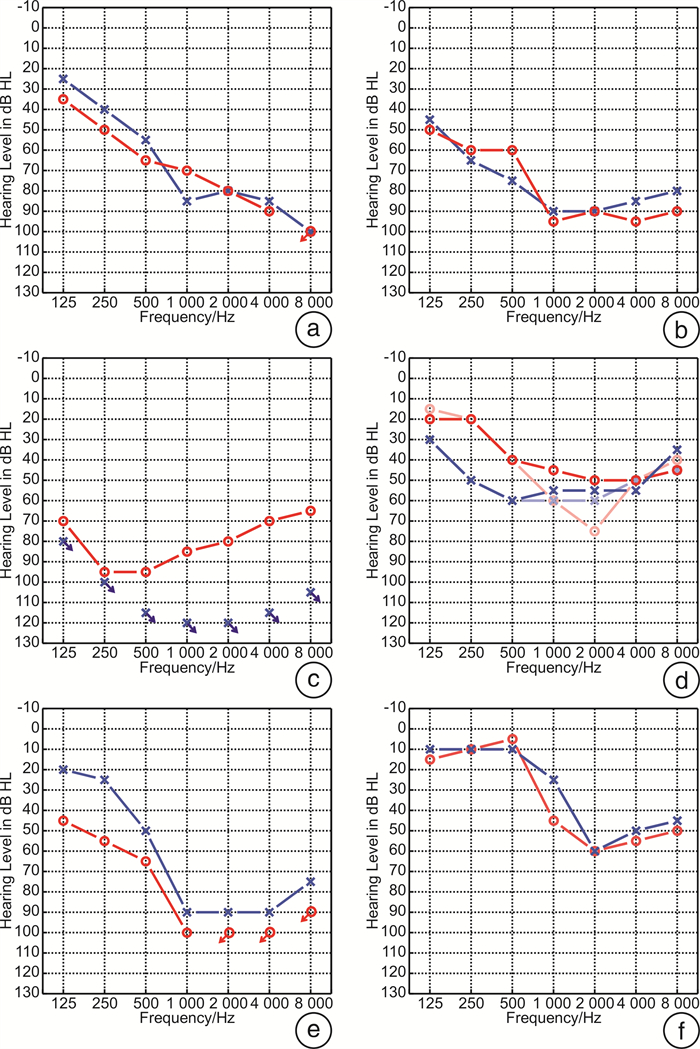
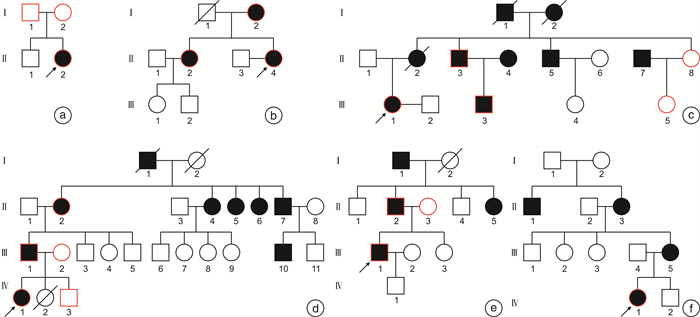
表 1 家系内成员听力损失首发年龄分布
家系 家系成员 首发年龄/岁 2108975 Ⅱ-1 10 1707828 Ⅱ-4 15 Ⅰ-1 50 Ⅱ-2 30 1808039 Ⅲ-1 20 Ⅰ-2 30~40 Ⅱ-3 18~19 1707748 Ⅳ-1 20 Ⅱ-2 30 Ⅲ-1 25 Ⅱ-4 30 Ⅱ-5 30 Ⅱ-7 35 1908345 Ⅲ-1 20 Ⅱ-2 17 Ⅱ-5 21 2108768 Ⅳ-1 17  下载: 导出CSV
下载: 导出CSV
表 2 先证者临床及听力学特征
家系 先证者 性别 检测年龄/岁 首发年龄/岁 病程/年 PTA-R/dB HL PTA-L/dB HL 听力损失程度 听力曲线类型 声导抗-R 声导抗-L 2108975 Ⅱ-1 女 25 10 15 76.00 76.00 重度 陡降型 Ad A 1707828 Ⅱ-4 女 39 15 24 87.50 85 极重度 陡降型 A A 1808039 Ⅲ-1 女 32 10 22 82.50 117.50 极重度 上升型 A A 1707748 Ⅳ-1 女 29 20 9 46.25 56.25 中度 谷型 A A 30 20 10 53.75 57.50 中重度 谷型 - - 1908345 Ⅲ-1 男 30 20 10 91.25 80.00 极重度 陡降型 A A 2108768 Ⅳ-1 女 23 17 6 41.00 36.00 中度 陡降型 As A Pbmax-R/% Pbmax-L/% ABR-R ABR-L DPOAE-R DPOAE-L - - - - - - - - - - - - - 阈值90 dBnHL,100 dBnHL可见Ⅲ、Ⅴ波 100 dBnHL未引出 各频率均未引出 各频率均未引出 68 84 100 dBnHLⅠ~Ⅴ间期正常 100 dBnHLⅠ~Ⅴ间期正常 各频率均未引出 各频率均未引出 - - - - - - 0 56 100 dBnHL未引出反应 100 dBnHL未引出反应 各频率均未引出 各频率均未引出 96 100 100 dBnHLⅠ~Ⅴ间期正常 100 dBnHLⅠ~Ⅴ间期正常 各频率均未引出 各频率均未引出 R:右耳,L:左耳,PTA:平均听阈,Pbmax:最大言语识别率,ABR:听性脑干反应,DPOAE:畸变产物耳声发射,-:无。
下载: 导出CSV
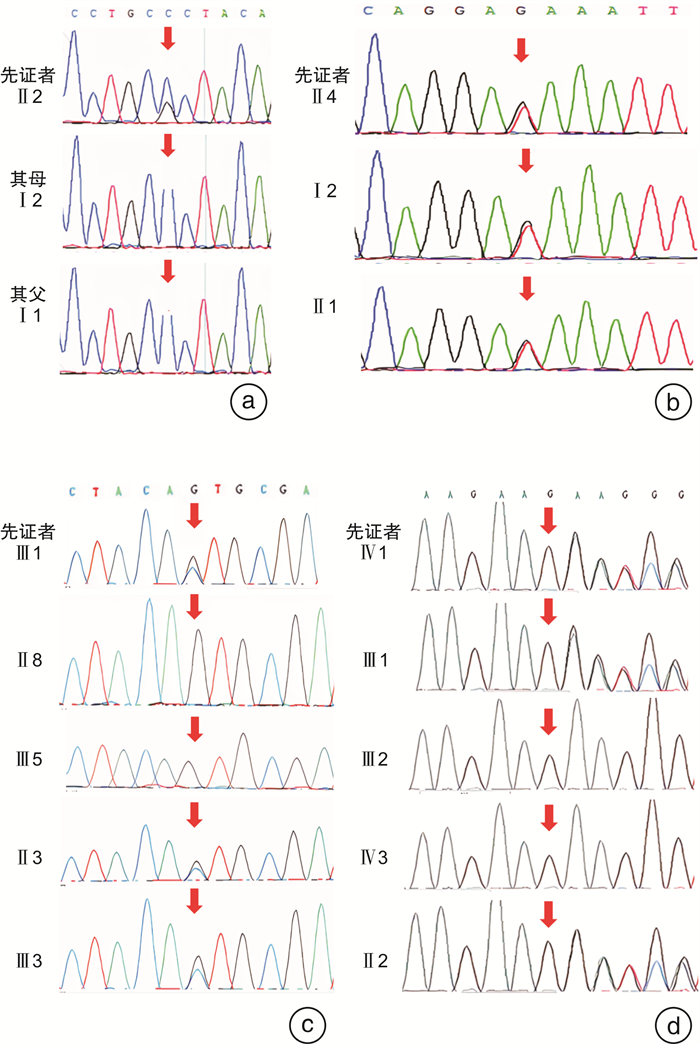
表 3 基因检测及致病性分析
家系 核苷酸
(NM_004403.2)绝对位置 变异类型 千人基因组 gnomAD数据库 ExAC数据库 致病性 致病变异证据
(2018)2108975 c.991-7C>G chr7:24,745,997-24,746,007(Intron 7) Splicing / African:0.000064,East Asian:0 / LP PS2+PM2 1707828 c.1183+1G>T chr7:24,745,797-24,745,807(Intron8) Splicing / / / LP PVS1+PM2+PP1 1808039 c.991-1G>C chr7:24,745,991-24,746,001(Intron7) Splicing / / / LP PVS1+PM2+PP1 1707748 c.991-15_991-13del chr7:24,746,003-24,746,01(Intron 7) Splicing / European:0.000043,East Asian:0 / P 1908345 2108768 P致病性,LP疑似致病性;PVS1非常强,PS2强,PM2中等,PP1辅助证据。
下载: 导出CSV
表 4 已报道的GSDME致病性变异
GSDME变异位点 RNA水平的影响 听力表型 国家 参考文献 碱基变异 变异位置 绝对位置 发病年龄/岁 听力损失类型 c.990+503_990+1691delins132 内含子7 chr7:24746055-24747243delins 第8外显子跳跃缺失 5~15 高频至全频 荷兰 Van Laer et al,1998[19] c.991-15_991-13del 内含子7 chr7:24746007-24746010 第8外显子跳跃缺失 7~30 高频 中国 Yu C et al,2003[20] 20 高频 韩国 Park HJ et al,2010[21] 10~30 高频 日本 Nishio A et al,2014[22] 18 高频 日本 Nishio A et al,2014[22] 6~20 全频 中国 Wang H et al,2018[8] 8~18 高频 东亚 Booth KT et al,2018[10] 10~15 高频 欧洲 Booth KT et al,2020[28] 25~35 高频 中国 Lei P et al,2022[29] c.991-6C>G 内含子7 chr7:24746001 第8外显子跳跃缺失 28~49 高频至全频 荷兰 Bischoff AM et al,2004[23] c.991-3C>A 内含子7 chr7:24745998 第8外显子跳跃缺失 20~39 高频至全频 中国 Wang H et al,2018[8] 20~40 高频至全频 中国 Chen,Xi et al,2020[24] c.991-2A>G 内含子7 chr7:24745997 第8外显子跳跃缺失 8~18 高频 中国 Chai Y et al,2014[25] >10 高频 欧洲 Booth KT et al,2018[10] 13~61 高频至全频 中国 Jin Z et al,2022[9] c.1183 G>C 外显子8 chr7:24745803 第8外显子跳跃缺失 中国 Chen S et al,2016[30] 18~25 高频至全频 中国 Li Q et al,2022[31] c.1154 C>T 外显子8 chr7:24745832 第8外显子跳跃缺失 >10 高频至全频 伊朗 Booth KT et al,2018[10] c.1102 C>G 外显子8 chr7:24745884 第8外显子跳跃缺失 >10 高频至全频 欧洲 Booth KT et al,2018[10] c.1183 G>A 外显子8 chr7:24745803 第8外显子跳跃缺失 >10 高频至全频 东亚 Booth KT et al,2018[10] IVS8+4 A>G 内含子8 chr7:24745799 第8外显子跳跃缺失 1~50 高频至全频 中国 Cheng J et al,2007[32] c.1183+1delG 内含子8 chr7:24745802 第8外显子跳跃缺失 8~30 高频至全频 中国 Li-Yang MN et al,2015[33] 12~30 高频至全频 中国 王诺扬等,2021[34] c.991-60_1095del 内含子7-外显子8 chr7:24745893-24746057 第8外显子跳跃缺失 4~59 高频至全频 法国 Mansard L et al,2022[35] c.[990+793_1007del;1029_1183+1376del] 内含子7-外显子8 chr7:24745979-24746953;chr7:24744430-24745960 第8外显子跳跃缺失 30 高频至全频 法国 Mansard L et al,2022[35]
下载: 导出CSV
-
[1] Clark JL, Swanepoel W. The World Report on Hearing-a new era for global hearing care[J]. Int J Audiol, 2021, 60(3): 161. doi: 10.1080/14992027.2021.1881318
[2] Cunningham LL, Tucci DL. Hearing Loss in Adults[J]. N Engl J Med, 2017, 377(25): 2465-2473. doi: 10.1056/NEJMra1616601
[3] van Camp G, Coucke P, Balemans W, et al. Localization of a gene for non-syndromic hearing loss(DFNA5) to chromosome 7p15[J]. Hum Mol Genet, 1995, 4(11): 2159-2163. doi: 10.1093/hmg/4.11.2159
[4] Thorpe RK, Walls WD, Corrigan R, et al. AudioGene: refining the natural history of KCNQ4, GSDME, WFS1, and COCH-associated hearing loss[J]. Hum Genet, 2022, 141(3-4): 877-887. doi: 10.1007/s00439-021-02424-7
[5] Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation[J]. Nat Rev Immunol, 2020, 20(3): 143-157. doi: 10.1038/s41577-019-0228-2
[6] Zhang Z, Zhang Y, Xia S, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity[J]. Nature, 2020, 579(7799): 415-420. doi: 10.1038/s41586-020-2071-9
[7] Wang Y, Gao W, Shi X, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin[J]. Nature, 2017, 547(7661): 99-103. doi: 10.1038/nature22393
[8] Wang H, Guan J, Guan L, et al. Further evidence for "gain-of-function" mechanism of DFNA5 related hearing loss[J]. Sci Rep, 2018, 8(1): 8424. doi: 10.1038/s41598-018-26554-7
[9] Jin Z, Zhu Q, Lu Y, et al. Identification of a novel DFNA5 mutation, IVS7-2 a>G, in a Chinese family with non-syndromic sensorineural hearing loss[J]. Acta Otolaryngol, 2022, 142(5): 448-453. doi: 10.1080/00016489.2019.1597984
[10] Booth KT, Azaiez H, Kahrizi K, et al. Exonic mutations and exon skipping: Lessons learned from DFNA5[J]. Hum Mutat, 2018, 39(3): 433-440. doi: 10.1002/humu.23384
[11] 关静, 贺林, 杨仕明, 等. 聋病遗传咨询专家共识[J]. 中华耳科学杂志, 2022, 20(2): 222-226. https://www.cnki.com.cn/Article/CJFDTOTAL-ZHER202202011.htm
[12] Chadha S, Kamenov K, Cieza A. The world report on hearing, 2021[J]. Bull World Health Organ, 2021, 99(4): 242-242A. doi: 10.2471/BLT.21.285643
[13] Oza AM, DiStefano MT, Hemphill SE, et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss[J]. Hum Mutat, 2018, 39(11): 1593-1613. doi: 10.1002/humu.23630
[14] Michaelson JJ, Shi Y, Gujral M, et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation[J]. Cell, 2012, 151(7): 1431-1442. doi: 10.1016/j.cell.2012.11.019
[15] Cabanillas R, Diñeiro M, Cifuentes GA, et al. Comprehensive genomic diagnosis of non-syndromic and syndromic hereditary hearing loss in Spanish patients[J]. BMC Med Genomics, 2018, 11(1): 58. doi: 10.1186/s12920-018-0375-5
[16] Guan J, Li J, Chen G, et al. Family trio-based sequencing in 404 sporadic bilateral hearing loss patients discovers recessive and De novo genetic variants in multiple ways[J]. Eur J Med Genet, 2021, 64(10): 104311. doi: 10.1016/j.ejmg.2021.104311
[17] Klimara MJ, Nishimura C, Wang D, et al. De novo variants are a common cause of genetic hearing loss[J]. Genet Med, 2022, 24(12): 2555-2567. doi: 10.1016/j.gim.2022.08.028
[18] Nadol JB Jr, Handzel O, Amr S. Histopathology of the Human Inner Ear in a Patient With Sensorineural Hearing Loss Caused by a Variant in DFNA5[J]. Otol Neurotol, 2015, 36(10): 1616-1621. doi: 10.1097/MAO.0000000000000888
[19] Van Laer L, Huizing EH, Verstreken M, et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5[J]. Nat Genet, 1998, 20(2): 194-197. doi: 10.1038/2503
[20] Yu C, Meng X, Zhang S, et al. A 3-nucleotide deletion in the polypyrimidine tract of intron 7 of the DFNA5 gene causes nonsyndromic hearing impairment in a Chinese family[J]. Genomics, 2003, 82(5): 575-579. doi: 10.1016/S0888-7543(03)00175-7
[21] Park HJ, Cho HJ, Baek JI, et al. Evidence for a founder mutation causing DFNA5 hearing loss in East Asians[J]. J Hum Genet, 2010, 55(1): 59-62. doi: 10.1038/jhg.2009.114
[22] Nishio A, Noguchi Y, Sato T, et al. A DFNA5 mutation identified in Japanese families with autosomal dominant hereditary hearing loss[J]. Ann Hum Genet, 2014, 78(2): 83-91. doi: 10.1111/ahg.12053
[23] Bischoff AM, Luijendijk MW, Huygen PL, et al. A novel mutation identified in the DFNA5 gene in a Dutch family: a clinical and genetic evaluation[J]. Audiol Neurootol, 2004, 9(1): 34-46. doi: 10.1159/000074185
[24] Chen X, Jia BL, Li MH, et al. Case Report: Novel Heterozygous DFNA5 Splicing Variant Responsible for Autosomal Dominant Non-syndromic Hearing Loss in a Chinese Family[J]. Front Genet, 2020, 11: 569284. doi: 10.3389/fgene.2020.569284
[25] Chai Y, Chen D, Wang X, et al. A novel splice site mutation in DFNA5 causes late-onset progressive non-syndromic hearing loss in a Chinese family[J]. Int J Pediatr Otorhinolaryngol, 2014, 78(8): 1265-1268. doi: 10.1016/j.ijporl.2014.05.007
[26] Rogers C, Fernandes-Alnemri T, Mayes L, et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death[J]. Nat Commun, 2017, 8: 14128. doi: 10.1038/ncomms14128
[27] Liao XX, Dai YZ, Zhao YZ, et al. Gasdermin E: A Prospective Target for Therapy of Diseases[J]. Front Pharmacol, 2022, 13: 855828. doi: 10.3389/fphar.2022.855828
[28] Booth KT, Azaiez H, Smith R. DFNA5(GSDME)c.991-15_991-13delTTC: Founder Mutation or Mutational Hotspot?[J]. Int J Mol Sci, 2020, 21(11): 3951. doi: 10.3390/ijms21113951
[29] Lei P, Zhu Q, Dong W, et al. Mutation analysis of the GSDME gene in a Chinese family with non-syndromic hearing loss[J]. PLoS One, 2022, 17(11): e0276233. doi: 10.1371/journal.pone.0276233
[30] Chen S, Dong C, Wang Q, et al. Targeted Next-Generation Sequencing Successfully Detects Causative Genes in Chinese Patients with Hereditary Hearing Loss[J]. Genet Test Mol Biomarkers, 2016, 20(11): 660-665. doi: 10.1089/gtmb.2016.0051
[31] Li Q, Wang S, Liang P, et al. A novel splice site variant c.1183+1G>C in DFNA5 causing autosomal dominant nonsyndromic hearing loss in a Chinese family[J]. BMC Med Genomics, 2022, 15(1): 163. doi: 10.1186/s12920-022-01315-8
[32] Cheng J, Han DY, Dai P, et al. A novel DFNA5 mutation, IVS8+4A>G, in the splice donor site of intron 8 causes late-onset non-syndromic hearing loss in a Chinese family[J]. Clin Genet, 2007, 72(5): 471-477. doi: 10.1111/j.1399-0004.2007.00889.x
[33] Li-Yang MN, Shen XF, Wei QJ, et al. IVS8+1 DelG, a Novel Splice Site Mutation Causing DFNA5 Deafness in a Chinese Family[J]. Chin Med J(Engl), 2015, 128(18): 2510-2515.
[34] 王诺扬, 陈灿明, 童鸣, 等. 一个迟发性耳聋家系的DFNA5基因变异分析[J]. 中华医学遗传学杂志, 2021, 38(2): 174-177.
[35] Mansard L, Vaché C, Bianchi J, et al. Identification of the First Single GSDME Exon 8 Structural Variants Associated with Autosomal Dominant Hearing Loss[J]. Diagnostics(Basel), 2022, 12(1): 207.
-
图(3)
表(4)
计量
- 文章访问数: 548
- PDF下载数: 209
- 施引文献: 0