
Identifications of the novel mutants on MYO7A in a family with non-syndromic hereditary deafness
-
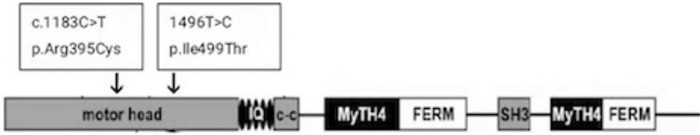
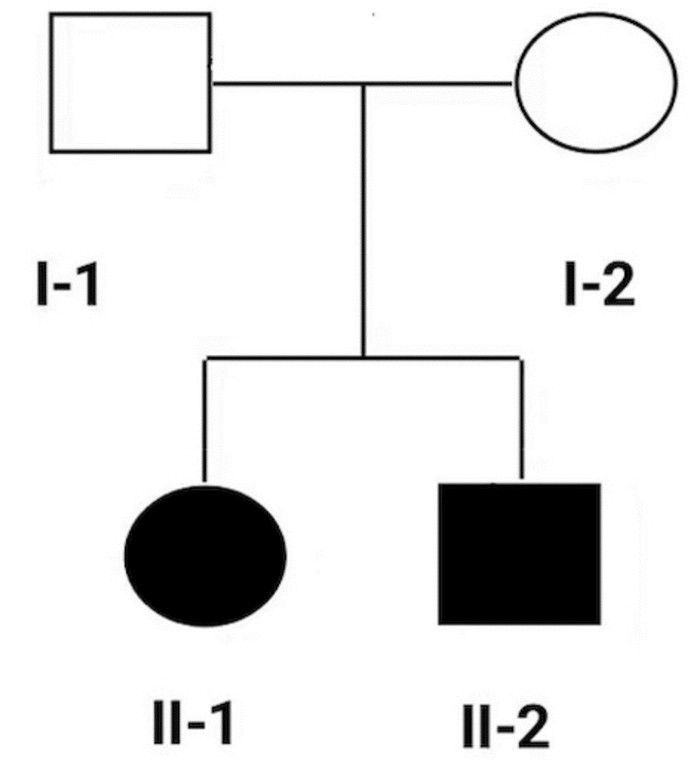
摘要: 目的 对一个非综合征性遗传性耳聋家系进行遗传学分析,查找该疾病的致聋性突变。方法 对该耳聋家系成员进行病史采集、听力、视力、基因组全外显子测序法检查分析,Sanger测序验证。结果 在 MYO7A 基因发现两个突变位点,分别为c.1183C>T、1496T>C,其中c.1183C>T有少量国外文献报道,1496T>C为新发现的突变位点。根据ACMG变异指南分析显示这两个突变为该先证者的致病突变。经Sanger测序分析验证,c.1183C>T来源于父亲,1496T>C来源于母亲。先证者及其弟弟听力检查结果均符合极重度感音神经性聋。在ESP6500数据库、千人基因数据库、Gnomad数据库中正常对照人群中均未发现这两个突变位点。结论 发现一个新的 MYO7A 基因致病性复合杂合突变,为 MYO7A 基因变异导致常染色体隐性遗传性非综合征性耳聋提供了更多的诊断依据,丰富了 MYO7A 基因突变谱,可提高对基因突变携带者高风险家庭的产前诊断率,减少出生缺陷。
-
关键词:
- MYO7A 基因 /
- 非综合征性遗传性耳聋 /
- 基因测序 /
- 突变检测
Abstract: Objective To identify the deaf-causing mutation by the genetic analysis in a family with non-syndromic hereditary deafness.Methods Medical history collection, hearing, vision, and genome whole-exome sequencing were performed on the members of the deaf family.Results Two mutation sites were identified in the MYO7A gene, namely c.1183C>T and 1496T>C, of which c.1183C>T has a small number of foreign literature reports, and 1496T>C is a newly discovered mutation site. According to ACMG mutation guideline showed that these two mutations were pathogenic mutations of the proband. Sanger sequencing verified that c.1183C>T was derived from the father, and 1496T>C was derived from the mother. These two mutation sites were not found in the healthy population in the Exome Sequencing Project(ESP6500) database, 1000 Genomes Project database, and the Gnomad database. Moreover, the second child in this family included a heterozygous mutation of c.1183C>T and 1496T>C and was confirmed to become severe sensorineural deaf.Conclusion A new pathogenic compound heterozygous mutation in the MYO7A gene has been discovered, which provides more diagnostic evidence for the autosomal recessive non-syndromic deafness caused by the MYO7A gene mutation and improves the prenatal gene diagnosis in high-risk families for mutation carriers to reduce congenital disabilities.-
Key words:
- MYO7A gene /
- non-syndromic hereditary deafness /
- DNA sequencing /
- mutation detection
-
-
表 1 家系基因型
基因 遗传方式 突变信息 Ⅰ-1 Ⅰ-2 Ⅱ-1 Ⅱ-2 MYO7A 常染色体隐性遗传 NM-000260.3:c.1183C>T(p.Arg395Cys) 杂合突变 无突变 杂合突变 杂合突变 MYO7A 常染色体隐性遗传 NM-000260.3:c.1496T>C(p.Ile499Thr) 无突变 杂合突变 杂合突变 杂合突变  下载: 导出CSV
下载: 导出CSV
-
[1] 周永安, 王湘, 马云霞, 等. 遗传性非综合征性常见耳聋基因诊断的研究[J]. 中国优生与遗传杂志, 2011, 19(7): 27-30, 39. https://www.cnki.com.cn/Article/CJFDTOTAL-ZYYA201107011.htm
[2] 张昊昱, 张宁, 张华, 等. 耳聋基因检测在遗传性耳聋诊断及遗传咨询中的应用[J]. 中华耳科学杂志, 2016, 14(5): 639-643. doi: 10.3969/j.issn.1672-2922.2016.05.017
[3] 孙艺, 袁慧军, 王荣光. MYO7A 基因突变与遗传性耳聋[J]. 中华耳科学杂志, 2010, 8(1): 63-67. doi: 10.3969/j.issn.1672-2922.2010.01.013
[4] Weil D, Blanchard S, Kaplan J, et al. Defective myosin VⅡA gene responsible for Usher syndrome type 1B[J]. Nature, 1995, 374(6517): 60-61. doi: 10.1038/374060a0
[5] Luijeindijk MWJ, van Wijk E, Bischoff AMLC, et al. Identific-ation and molecular modelling of a mutation in the motor head domain of myosin VⅡA in a family with autosomal dominant hearing impairment(DFNA11)[J]. Hum Genet, 2004, 115(2): 149-156. doi: 10.1007/s00439-004-1137-3
[6] Wolfrum U, Nagel-Wolfrum K. [The Usher Syndrome, a Human Ciliopathy][J]. Klin Monbl Augenheilkd, 2018, 235(3): 273-280. doi: 10.1055/a-0573-9431
[7] Boëda B, El-Amraoui A, Bahloul A, et al. Myosin VⅡa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle[J]. EMBO J, 2002, 21(24): 6689-6699. doi: 10.1093/emboj/cdf689
[8] 张会敏, 陈森, 孙宇, 等. 聋病基因诊断在评估人工耳蜗植入术预后中的价值[J]. 临床耳鼻咽喉头颈外科杂志, 2021, 35(3): 274-281. https://www.cnki.com.cn/Article/CJFDTOTAL-LCEH202103021.htm
[9] 郭敏, 韩炜伟, 李书聆. Usher1C新发突变致先天性感音神经性聋1例并文献复习[J]. 临床耳鼻咽喉头颈外科杂志, 2020, 34(6): 562-564. https://www.cnki.com.cn/Article/CJFDTOTAL-LCEH202006020.htm
[10] Astuto LM, Kelley PM, Askew JW, et al. Searching for evidence of DFNB2[J]. Am J Med Genet, 2002, 109(4): 291-297. doi: 10.1002/ajmg.10384
[11] Su MC, Yang JJ, Su CC, et al. Identification of novel variants in the Myosin VⅡA gene of patients with nonsyndromic hearing loss from Taiwan[J]. Int J Pediatr Otorhinolaryngol, 2009, 73(6): 811-815. doi: 10.1016/j.ijporl.2009.02.009
[12] Naz S, Imtiaz A, Mujtaba G, et al. Genetic causes of moderate to severe hearing loss point to modifiers[J]. Clin Genet, 2017, 91(4): 589-598. doi: 10.1111/cge.12856
[13] Shahzad M, Sivakumaran TA, Qaiser TA, et al. Genetic analysis through OtoSeq of Pakistani families segregating prelingual hearing loss[J]. Otolaryngol Head Neck Surg, 2013, 149(3): 478-487. doi: 10.1177/0194599813493075
[14] Richard EM, Santos-Cortez R, Faridi R, et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss[J]. Hum Mutat, 2019, 40(1): 53-72. doi: 10.1002/humu.23666
-



图(5)
表(1)
计量
- 文章访问数: 1996
- PDF下载数: 1861
- 施引文献: 0