
Analysis of molecular genetics and clinical characteristics of 3 children with Waardenburg syndrome
-
摘要: 目的 对3例综合征性聋患儿进行分子遗传学及临床特征分析,明确其致病基因及遗传特征。方法 对3例患儿及其父母进行病历采集和整理,包括常规检查和听力评估及颞骨CT、头颅MR检查;通过外显子组测序(WES)筛查可能致病的基因变异,并使用Sanger测序对先证者及其父母进行候选阳性变异验证。结果 3例患儿均为智力正常女性,例2患儿为散发,其余均有耳聋家族史,并符合常染色体显性遗传规律。3例患儿均为双侧极重度感音神经性听力障碍伴眼睛亮蓝色巩膜。其他表型包括眼距增宽(例1)、多发性色素异常沉着(例2)和毛发微黄(例2)、眼睑下垂(例3)。例3患儿影像学检查示双侧前庭扩大、内听道扩大,及双侧内耳畸形。例1患儿母亲仅有左侧轻度听力障碍;例3患儿母亲为双侧听力障碍,伴有单侧亮蓝色巩膜及毛发微黄。WES检出例1、例2、例3患儿分别有不同基因的杂合性变异:PAX3 c.811C>T、MITF c.632T>C,及SOX10 c.1359_1360 insGCCCCACA。例1和例3患儿检出变异均遗传自有听力障碍的母亲,例2患儿可能为自发变异。最终诊断例1患儿为Waardenburg综合征1型(WS1),例1患儿母亲、例2患儿、例3患儿及例3患儿母亲均为WS2。结论 WS为综合征性聋,临床诊断所依据的主要特征包括常染色体显性遗传和巩膜色素异常。研究表明,同一基因变异导致的WS仍存在表型异质性,其中PAX3变异导致轻度的WS则很可能是外显不全所致,因此,WS的确诊有赖于基因检测结果。例1、例3患儿突变位点为首次在患者中发现并鉴定,扩展了WS的致病性变异谱。
-
关键词:
- 综合征性聋 /
- Waardenburg综合征 /
- 分子遗传学 /
- 临床特征
Abstract: Objective To analyze the molecular genetics and clinical characteristics of 3 children with syndromic deafness were analyzed to clarify their causative genes and genetic characteristics.Methods The medical records of 3 children and their parents were collected and analyzed, including physical examination, hearing evaluation, temporal bone CT, and cranial MRI. Whole-exome sequencing(WES) was used to screen for pathogenic gene variants, and Sanger sequencing was used to verify the candidate positive variants in the probands and their parents.Results All 3 patients were female with normal intelligence. Patient 1 and 3 had a family history of deafness, which conformed to the pattern of autosomal dominant inheritance. All three patients had bilateral profound sensorineural hearing impairment with bright-blue sclera. Other phenotypes included hypertelorism(patient 1), multiple dyschromatosis(patient 2), and yellowish hair(patient 2), blepharoptosis(patient 3). Patient 3 had bilateral vestibular enlargement, internal auditory canal enlargement, and bilateral inner ear malformations. Mother of patient 1 had only left mild hearing impairment; mother of patient 3 had bilateral hearing impairment with unilateral bright-blue sclera and yellowish hair. WES detected heterozygous variants, PAX3 c.811C>T, MITF c.632T>C, and SOX10 c.1359_1360 insGCCCCACA, in patient 1, 2, and 3, respectively. The variants in patient 1 and 3 were inherited from their mothers who had hearing impairment, and MITFvariant in patient 2 may be a spontaneous variation. The final diagnoses were that patient 1 with Waardenburg syndrome type 1(WS1), and the mother of patient 1, patient 2, patient 3, and the mother of patient 3 with WS2.Conclusion WS is a syndromic deafness, and the main clinical features include autosomal dominant inheritance and scleral pigment abnormalities. However, the findings of this study show that there is still phenotypic heterogeneity in WS even caused by the same gene variant, so it depends on genetic tests to confirm the diagnosis; The gene variant of patient 1 and 2 was never been reported in other patients, which expands the pathogenic variant spectrum of WS.-
Key words:
- syndromic deafness /
- Waardenburg syndrome /
- molecular genetics /
- clinical features
-
-
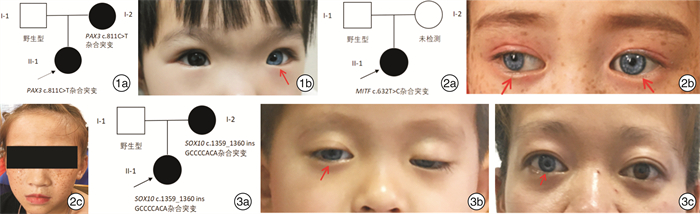
图 1 例1患儿家系图和临床表现 1a: 家系图; 1b: 先证者1左眼呈现蓝巩膜(红色箭头指示); 图 2 例2患儿家系图及临床表现 2a: 家系图; 2b: 先证者2双眼呈现蓝色巩膜(红色箭头指示); 2c: 先证者2满脸雀斑; 图 3 例3患儿家系图及临床表现 3a: 家系图; 3b: 先证者3右眼呈现蓝色巩膜(红色箭头所示), 左上睑下垂; 3c: 先证者3母亲右眼蓝色巩膜(红色箭头所示)。
表 1 3例广西WS患儿临床特征和分子诊断结果
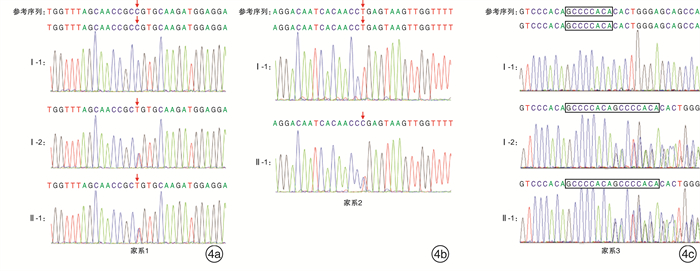
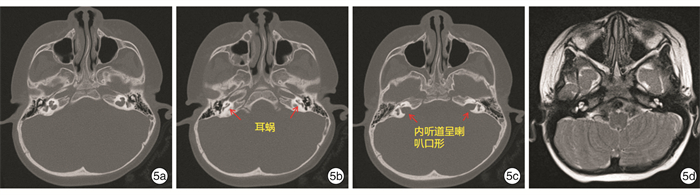
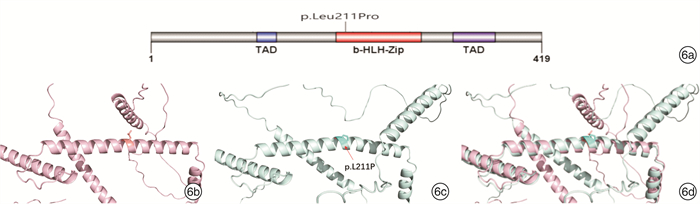
项目 例1患儿 例2患儿 例3患儿 性别 女 女 女 诊断年龄 1岁4个月 10岁10个月 2岁8个月 民族 汉族 瑶族 汉族 体检 左侧亮蓝色巩膜,眼距增宽 双侧亮蓝色巩膜,面部雀斑,毛发微黄 右侧亮蓝色巩膜,左上睑下垂 ABR/dB nHL 双侧>95 双侧>98 双侧>95 DPOAE 未引出 未引出 未引出 ASSR/dB nHL 双侧均>95 双侧均>100 左>97,右>95 影像学检查 未见明显异常 未见明显异常 颞骨CT提示双侧前庭扩大,内听道扩大,头颅MRI提示双侧内耳畸形 家族史 母亲左侧轻度听力障碍 无 母亲双侧极重度感音神经性聋,右侧亮蓝色巩膜,发色微黄 报告基因 PAX3 MITF SOX10 核酸改变 c.811C>T c.632T>C c.1359_1360ins GCCCCACA 氨基酸改变 p.Arg271Cys p.Leu211Pro p.His454Alafs 最终诊断 WS1 WS2 WS2  下载: 导出CSV
下载: 导出CSV
-
[1] Pingault V, Ente D, Dastot-Le MF, et al. Review and update of mutations causing Waardenburg syndrome[J]. Hum Mutat, 2010, 31(4): 391-406. doi: 10.1002/humu.21211
[2] Song J, Feng Y, Acke FR, et al. Hearing loss in Waardenburg syndrome: a systematic review[J]. Clin Genet, 2016, 89(4): 416-425. doi: 10.1111/cge.12631
[3] Huang S, Song J, He C, et al. Genetic insights, disease mechanisms, and biological therapeutics for Waardenburg syndrome[J]. Gene Ther, 2022, 29(9): 479-497. doi: 10.1038/s41434-021-00240-2
[4] 李霞, 赵声波, 毕先云, 等. PAX3基因新突变致Ⅰ型Waardenburg综合征家系基因型与表型特征分析[J]. 临床耳鼻咽喉头颈外科杂志, 2021, 35(7): 621-626. https://lceh.cbpt.cnki.net/WKC/WebPublication/paperDigest.aspx?paperID=bf972eab-9608-413b-a5ae-a75014886d14
[5] Somashekar PH, Upadhyai P, Narayanan DL, et al. Phenotypic diversity and genetic complexity of PAX3-related Waardenburg syndrome[J]. Am J Med Genet A, 2020, 182(12): 2951-2958. doi: 10.1002/ajmg.a.61893
[6] Wang J, Lu Y, Yan X, et al. Identification of novel MITF mutations in Chinese families with Waardenburg syndrome type Ⅱ[J]. Mol Genet Genom Med, 2021, 9(9): e1770.
[7] Pingault V, Zerad L, Bertani-Torres W, et al. SOX10: 20 years of phenotypic plurality and current understanding of its developmental function[J]. J Med Genet, 2022, 59(2): 105-114.
[8] Fleck K, Erhardt G, Luhken G. From single nucleotide substitutions up to chromosomal deletions: genetic pause of leucism-associated disorders in animals[J]. Berl Munch Tierarztl Wochenschr, 2016, 129(7/8): 269-281.
[9] 马静, 明澄, 林垦, 等. Ⅱ型Waardenburg综合征患儿二例基因诊断分析[J]. 中华耳鼻咽喉头颈外科杂志, 2021, 56(1): 47-54.
[10] 徐彬, 戴继任, 毕静, 等. 新一代测序技术在3例Waardenburg综合征患儿中的应用[J]. 临床耳鼻咽喉头颈外科杂志, 2021, 35(10): 910-913. https://lceh.cbpt.cnki.net/WKC/WebPublication/paperDigest.aspx?paperID=28dcb878-8fb4-4909-8878-60a553cee0c0
[11] Somashekar PH, Girisha KM, Nampoothiri S, et al. Locus and allelic heterogeneity and phenotypic variability in Waardenburg syndrome[J]. Clin Genet, 2019, 95(3): 398-402.
[12] Boudjadi S, Chatterjee B, Sun W, et al. The expression and function of PAX3 in development and disease[J]. Gene, 2018, 666: 145-157.
[13] Pingault V, Ente D, Dastot-Le MF, et al. Review and update of mutations causing Waardenburg syndrome[J]. Hum Mutat, 2010, 31(4): 391-406.
[14] Birrane G, Soni A, Ladias JA. Structural basis for DNA recognition by the human PAX3 homeodomain[J]. Biochemistry, 2009, 48(6): 1148-1155.
[15] Chi YI. Homeodomain revisited: a lesson from disease-causing mutations[J]. Hum Genet, 2005, 116(6): 433-444.
[16] Lang D, Epstein JA. Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer[J]. Hum Mol Genet, 2003, 12(8): 937-945.
[17] Alehabib E, Alinaghi S, Pourfatemi F, et al. Incomplete penetrance of MITF gene c. 943C > T mutation in an extended family with Waardenburg syndrome type Ⅱ[J]. Int J Pediatr Otorhinolaryngol, 2020, 135: 110014.
[18] Grill C, Bergsteinsdottir K, Ogmundsdottir MH, et al. MITF mutations associated with pigment deficiency syndromes and melanoma have different effects on protein function[J]. Hum Mol Genet, 2013, 22(21): 4357-4367.
[19] Brownstein Z, Gulsuner S, Walsh T, et al. Spectrum of genes for inherited hearing loss in the Israeli Jewish population, including the novel human deafness gene ATOH1[J]. Clin Genet, 2020, 98(4): 353-364.
[20] Tachibana M, Takeda K, Nobukuni Y, et al. Ectopic expression of MITF, a gene for Waardenburg syndrome type 2, converts fibroblasts to cells with melanocyte characteristics[J]. Nat Genet, 1996, 14(1): 50-54.
[21] Thongpradit S, Jinawath N, Javed A, et al. Novel SOX10 Mutations in Waardenburg Syndrome: Functional Characterization and Genotype-Phenotype Analysis[J]. Front Genet, 2020, 11: 589784.
[22] Chaoui A, Watanabe Y, Touraine R, et al. Identification and functional analysis of SOX10 missense mutations in different subtypes of Waardenburg syndrome[J]. Hum Mutat, 2011, 32(12): 1436-1449.
[23] Elmaleh-Berges M, Baumann C, Noel-Petroff N, et al. Spectrum of temporal bone abnormalities in patients with Waardenburg syndrome and SOX10 mutations[J]. AJNR Am J Neuroradiol, 2013, 34(6): 1257-1263.
[24] 徐广雨, 郝青青, 钟玲玲, 等. SOX10基因突变对Waardenburg综合征患者内耳发育的影响[J]. 中华耳鼻咽喉头颈外科杂志, 2016, 51(11): 832-837. https://cdmd.cnki.com.cn/Article/CDMD-90115-1016235019.htm
-
图(4)
表(1)
计量
- 文章访问数: 1463
- PDF下载数: 168
- 施引文献: 0