
The genotype-phenotype correlation analysis and genetic counseling of hearing loss patients with novel KCNQ4 mutations
-
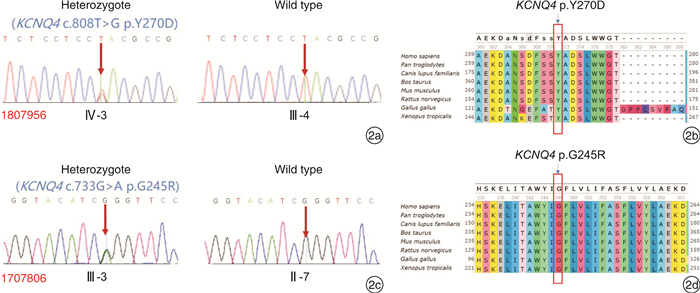
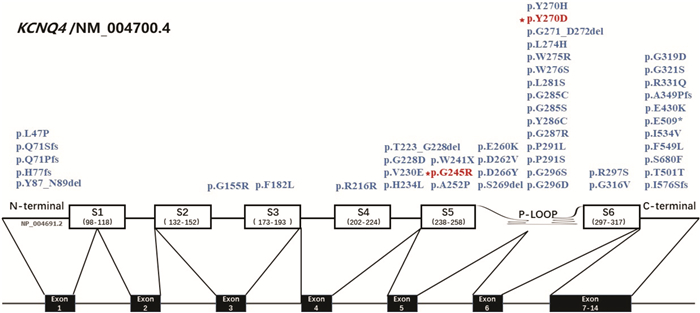
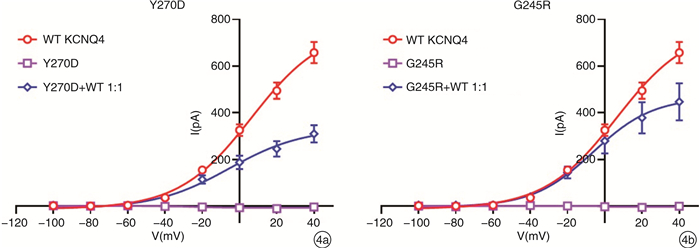
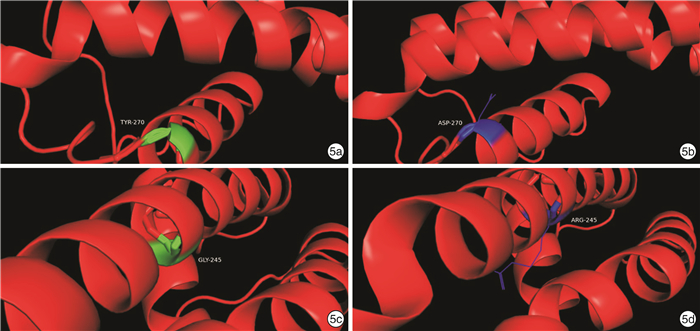
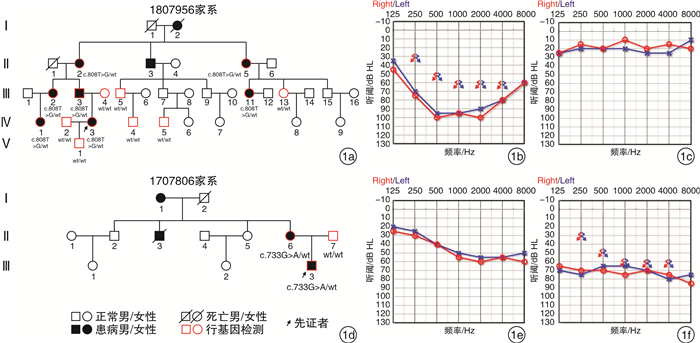
摘要: 目的 对KCNQ4基因新突变耳聋患者进行基因型表型相关性分析,为患者及家庭提供精准的遗传咨询。方法 纳入1807956(5代34人)和1707806(3代12人)两个耳聋大家系,应用二代测序技术检测先证者致病基因,并对家系内成员进行Sanger测序验证。根据美国医学遗传学与基因组学会(ACMG)指南,结合临床资料、基因检测、生物信息分析、电生理实验等对突变位点进行致病性分析,为患者提供遗传咨询。结果 1807956家系先证者为育龄女性,15岁发病,携带KCNQ4 c.808T>G p.Y270D变异,双耳极重度听力下降,中频听力下降更为明显;1707806家系先证者为青少年,11岁发病,携带KCNQ4 c.733G>A p.G245R变异,双耳中重度听力下降,以高频为主。两个家系每一代均有患者,患者无性别差异,双亲之一也为患者,符合常染色体显性遗传发病特点。两个变异均为错义突变,在家系中共分离,正常人群中无分布。生物信息分析工具预测变异有害,变异位点在不同物种中高度保守,电生理实验提示变异离子通道功能受损。根据ACMG指南,判定KCNQ4 c.808T>G变异为致病的,KCNQ4 c.733G>A变异为可能致病的。结论 新发现的两个突变位点为国际首次报道,患者听力下降特点具有异质性,丰富了KCNQ4基因变异谱及临床表型,为患者及家庭提供了可靠的遗传咨询。Abstract: Objective To provide accurate genetic counseling, the genotype-phenotype correlation of the patients with KCNQ4 mutations was analyzed.Methods Two hearing loss families, 1807956(a five-generation family with 34 members) and 1707806(a three-generation family with 12 members) were recruited. The candidate variants were detected by next generation sequencing technology. Sanger sequencing was performed to verify the co-segregation of the phenotype in the recruited family members. According to American College of Medical Genetics and Genomics(ACMG) guideline, combined with clinical data, genetic testing, bioinformatic analysis and electrophysiological experiments, the pathogenicity of mutations was analyzed and genetic counseling was provided for family members.Results The proband of family 1807956 was a pregnant woman, who carried KCNQ4 c.808T>G p.Y270D and developed hearing loss at the age of 15 years old, she had profound hearing loss in both ears, with middle-frequency highly affected. The proband of family 1707806 was an adolescent whose onset age was 11 years old, carrying KCNQ4 c.733G>A p.G245R, he presented with bilateral moderately severe hearing loss. The inheritance pattern of these two families were autosomal dominant inheritance. The two variants were missense mutations that were co-segregation in the two families and were not found in normal population. The mutations predicted by bioinformatic analysis tools were damaging and highly conserved in different species. Electrophysiological experiments showed that the function of the mutant ion channels was impaired. According to ACMG guideline, KCNQ4 c.808T>G was pathogenic, and KCNQ4 c.733G>A was likely pathogenic.Conclusion The two mutations in this research were reported for the first time. The hearing loss of the patients showed heterogeneity, enriching the variation spectrum and clinical phenotype of KCNQ4.
-
Key words:
- genetic hearing loss /
- KCNQ4 /
- autosomal dominant /
- genetic counseling
-
-
表 1 本研究KCNQ4基因变异位点致病性分析
变异位点 软件预测 最小等位基因频率 Clin Var DVD ACMG致病变异分级标准 变异类型 PROVEAN SIFT PolyPhen-2 Mutation Taster CADD Phred 1000 Genomes ExAC KCNQ4 c.808T>G p.Y270D 有害 有害 可能有害 致病的 28.5 0 0 - - PS3、PP1_ Strong、PM1、PM2、PM5、PP3 致病 KCNQ4 c.733G>A p.G245R 有害 有害 可能有害 致病的 31 0 0 - 意义未明 PS3、PM2,PP1、PP3 可能致病 PS3:体内、体外功能实验已明确会导致基因功能受损的变异;PM1:位于热点突变区域和/或位于已知无良性变异的关键功能域;PM2:ESP数据库、千人数据库、EXAC数据库中正常对照人群中未发现的变异;PM5:新的错义突变导致氨基酸变化,此变异之前未曾报道,但是在同一位点,导致另外一种氨基酸的变异已经确认是致病性的;PP1:突变与疾病在家系中共分离;PP3:多种统计方法预测出该变异会对基因或基因产物造成有害的影响,包括保守性预测、进化预测、剪接位点影响等。  下载: 导出CSV
下载: 导出CSV
-
[1] Del Castillo I, Morín M, Domínguez-Ruiz M, et al. Genetic etiology of non-syndromic hearing loss in Europe[J]. Hum Genet, 2022, 141(3/4): 683-696.
[2] Sloan-Heggen CM, Bierer AO, Shearer AE, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss[J]. Hum Genet, 2016, 135(4): 441-450. doi: 10.1007/s00439-016-1648-8
[3] Naito T, Nishio SY, Iwasa Y, et al. Comprehensive genetic screening of KCNQ4 in a large autosomal dominant nonsyndromic hearing loss cohort: genotype-phenotype correlations and a founder mutation[J]. PLoS One, 2013, 8(5): e63231. doi: 10.1371/journal.pone.0063231
[4] Chadha S, Kamenov K, Cieza A. The world report on hearing, 2021[J]. Bull World Health Organ, 2021, 99(4): 242-242A. doi: 10.2471/BLT.21.285643
[5] Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5): 405-424. doi: 10.1038/gim.2015.30
[6] Oza AM, DiStefano MT, Hemphill SE, et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss[J]. Hum Mutat, 2018, 39(11): 1593-1613. doi: 10.1002/humu.23630
[7] Kubisch C, Schroeder BC, Friedrich T, et al. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness[J]. Cell, 1999, 96(3): 437-446. doi: 10.1016/S0092-8674(00)80556-5
[8] Carignano C, Barila EP, Rías EI, et al. Inner Hair Cell and Neuron Degeneration Contribute to Hearing Loss in a DFNA2-Like Mouse Model[J]. Neuroscience, 2019, 410: 202-216. doi: 10.1016/j.neuroscience.2019.05.012
[9] Michalski N, Petit C. Genes Involved in the Development and Physiology of Both the Peripheral and Central Auditory Systems[J]. Annu Rev Neurosci, 2019, 42: 67-86. doi: 10.1146/annurev-neuro-070918-050428
[10] 牛文侠, 许慧娟, 秦利涛, 等. 42例迟发性非综合征性耳聋患者基因型与临床表型分析[J]. 临床耳鼻咽喉头颈外科杂志, 2021, 35(2): 131-136. https://lceh.cbpt.cnki.net/WKC/WebPublication/paperDigest.aspx?paperID=cd1c475c-9007-435e-a459-1d2203652404
[11] 张秋静, 吴楷文, 赵翠, 等. 中国迟发性非综合征性耳聋家系CEACAM16基因新突变的鉴定[J]. 临床耳鼻咽喉头颈外科杂志, 2020, 34(9): 777-781. https://lceh.cbpt.cnki.net/WKC/WebPublication/paperDigest.aspx?paperID=70ebefc8-fc4b-44b8-bec0-6444451094d2
[12] Shin DH, Jung J, Koh YI, et al. A recurrent mutation in KCNQ4 in Korean families with nonsyndromic hearing loss and rescue of the channel activity by KCNQ activators[J]. Hum Mutat, 2019, 40(3): 335-346.
[13] Jung J, Choi HB, Koh YI, et al. Whole-exome sequencing identifies two novel mutations in KCNQ4 in individuals with nonsyndromic hearing loss[J]. Sci Rep, 2018, 8(1): 16659. doi: 10.1038/s41598-018-34876-9
[14] Namba K, Mutai H, Kaneko H, et al. In silico modeling of the pore region of a KCNQ4 missense mutant from a patient with hearing loss[J]. BMC Res Notes, 2012, 5: 145. doi: 10.1186/1756-0500-5-145
[15] Shen J, Oza AM, Del Castillo I, et al. Consensus interpretation of the p. Met34Thr and p. Val37Ile variants in GJB2 by the ClinGen Hearing Loss Expert Panel[J]. Genet Med, 2019, 21(11): 2442-2452.
[16] Thorpe RK, Walls WD, Corrigan R, et al. AudioGene: refining the natural history of KCNQ4, GSDME, WFS1, and COCH-associated hearing loss[J]. Hum Genet, 2022, 141(3/4): 877-887.
[17] Rim JH, Choi JY, Jung J, et al. Activation of KCNQ4 as a Therapeutic Strategy to Treat Hearing Loss[J]. Int J Mol Sci, 2021, 22(5): 2510. doi: 10.3390/ijms22052510
[18] Gao X, Tao Y, Lamas V, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents[J]. Nature, 2018, 553(7687): 217-221. doi: 10.1038/nature25164
-
图(5)
表(1)
计量
- 文章访问数: 2346
- PDF下载数: 705
- 施引文献: 0