
Genotype and phenotype analysis of a family with Waardenburg syndrome type Ⅰcaused by a novel mutation in PAX3 gene
-
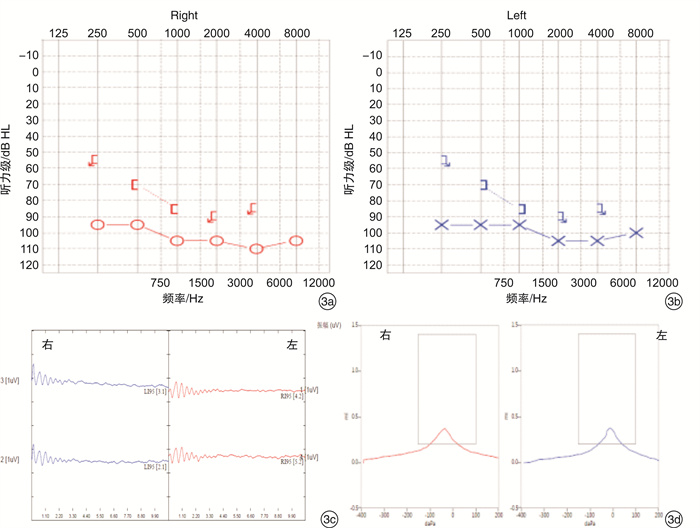
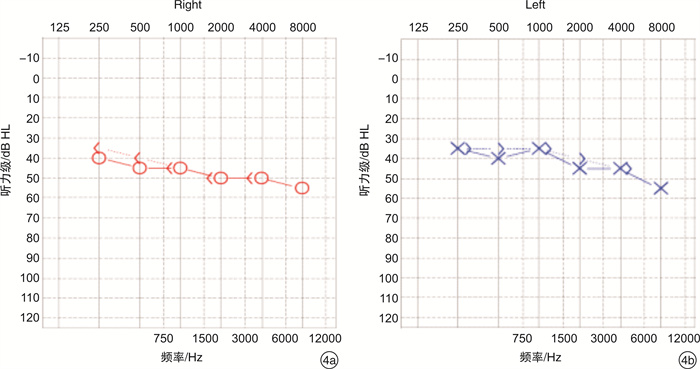
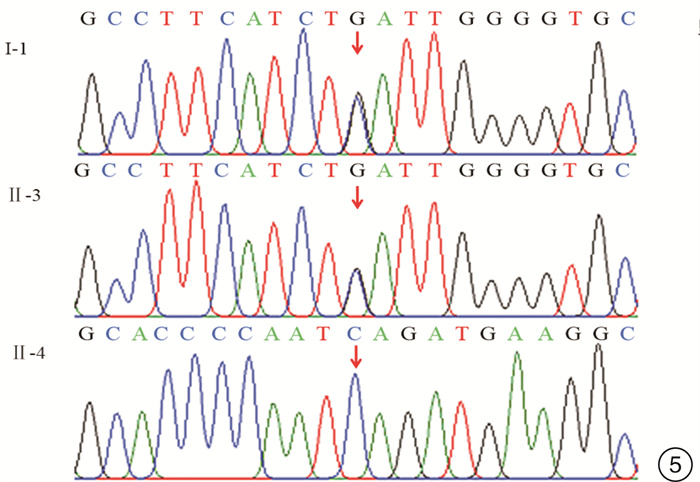
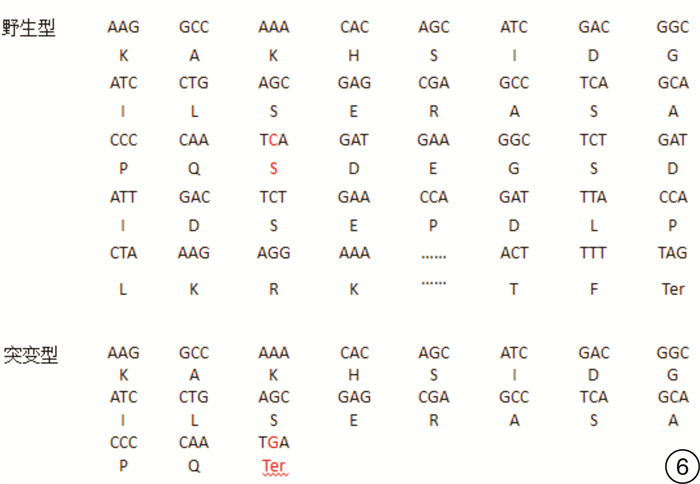
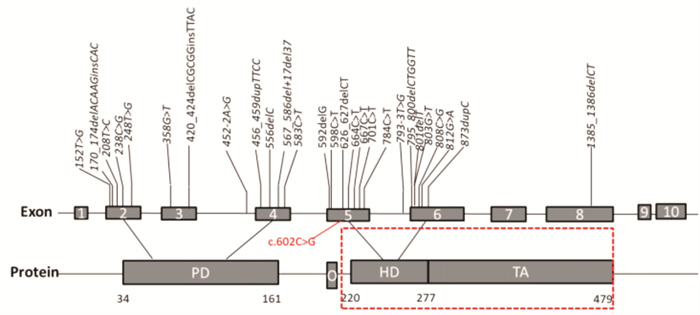
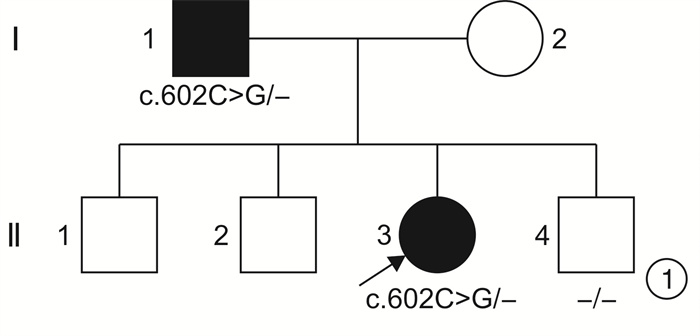
摘要: 目的 通过对云南地区Ⅰ型Waardenburg综合征(WS)一家系的突变基因致病性进行鉴定分析,探讨可能的分子生物学致病原因。方法 经知情同意,对具有WS表型的先证者及其家属进行病史采集、体格检查、听力学评估。获取外周血,提取基因组DNA,高通量测序方法对耳聋相关基因进行检测,对先证者及其家属进行突变位点的Sanger测序验证分析。结果 先证者高通量测序发现 PAX3 基因第5外显子c.602C>G突变,该突变为无义突变。导致编码蛋白质第201位氨基酸由丝氨酸变为终止密码子,氨基酸提前终止翻译,蛋白质截短。经Sanger测序验证先证者父亲携带相同位点的突变,弟弟该位点未突变。根据美国医学遗传学与基因组学学会遗传变异分类标准与指南(ACMG),判定为致病性(PVS1+PM2+PP3)。保守性分析提示多个物种氨基酸序列一致,具有高度保守性。结论 结合临床诊断及基因诊断结果,初步认定该突变为患儿致病原因。本研究丰富了 PAX3 基因的突变图谱,为临床分子诊断及遗传咨询提供了一定参考。
-
关键词:
- 耳聋基因 /
- Waardenburg综合征 /
- PAX3 /
- 基因型 /
- DNA突变分析
Abstract: Objective To identify gene mutation and analysis the association between clinical characterizes and the mutations in a family of Waardenburg syndrome (WS) type I in Yunnan, China.Methods With informed consent, the proband with WS phenotype and his family members were given medical history collection, physical examination and audiological evaluation. Peripheral blood was obtained, genomic DNA was extracted, and deafness related genes were detected by high-throughput sequencing. Sanger sequencing was used to verify the mutation sites of proband and his family members.Results C. 602C>G mutation in exon 5 of PAX3 gene was identified, which is nonsense mutation and may cause a truncated protein. The mutation cause 201 amino acid of the protein changed from serine to stop codon. According to the American College of Medical Genetics and Genomics (ACMG), it is considered as Pathogenicity(PVS1+PM2+PP3). This mutation has not been included in the database also not been reported in the literature.Conclusion Combined with the results of clinical diagnosis and gene diagnosis, this mutation was considered as the cause of the disease. This study enriched mutation spectrum of PAX3 gene.-
Key words:
- deafness gene /
- Waardenburg syndrome /
- PAX3 /
- genotype /
- DNA mutational analysis
-
-
[1] Waardenburg PJ.A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness[J]. Am J Hum Genet, 1951, 3(3):195-253.
[2] Yu Y, Liu W, Chen M, et al.Two novel mutations of PAX3 and SOX10 were characterized as genetic causes of Waardenburg Syndrome[J]. Mol Genet Genomic Med, 2020, 8(5):e1217.
[3] Pingault V, Ente D, Dastot-Le Moal F, et al.Review and update of mutations causing Waardenburg syndrome[J]. Hum Mutat, 2010, 31(4):391-406. doi: 10.1002/humu.21211
[4] 马静, 明澄, 林垦, 等.Ⅱ型Waardenburg综合征患儿二例基因诊断分析[J].中华耳鼻咽喉头颈外科杂志, 2021, 56(1):4754.
[5] Ma J, Lin K, Jiang HC, et al.A novel mutation of the PAX3 gene in a Chinese family with Waardenburg syndrome type I[J]. Mol Genet Genomic Med, 2019, 7(7):e00798.
[6] 刘梦婷, 张天虹.综合征型耳聋的诊断与治疗策略[J].临床耳鼻咽喉头颈外科杂志, 2021, 35(3):285-288. https://www.cnki.com.cn/Article/CJFDTOTAL-LCEH202103023.htm
[7] Huang S, Song J, He C, et al.Genetic insights, disease mechanisms, and biological therapeutics for Waardenburg syndrome[J]. Gene Ther, 2021, 25.
[8] Kim H, Ankamreddy H, Lee DJ, et al.Pax3 function is required specifically for inner ear structures with melanogenic fates[J]. Biochem Biophys Res Commun, 2014,445(3):608-614. doi: 10.1016/j.bbrc.2014.02.047
[9] Vogan KJ, Underhill DA, Gros P.An alternative splicing event in the Pax-3 paired domain identifies the linker region as a key determinant of paired domain DNA-binding activity[J]. Mol Cell Biol, 1996, 16(12):6677-6686. doi: 10.1128/MCB.16.12.6677
[10] Boudjadi S, Chatterjee B, Sun W, et al.The expression and function of PAX3 in development and disease[J]. Gene, 2018,666:145-157. doi: 10.1016/j.gene.2018.04.087
[11] 王晶, 赵娜, 薛煜, 等.全外显子测序技术在Waardenburg综合征家系致病基因突变分析中的应用[J].山西大学学报(自然科学版), 2018, 41(3):656-660. https://www.cnki.com.cn/Article/CJFDTOTAL-SXDR201803029.htm
[12] 郝子琪, 周永安, 李鹏丽, 等.七例Waardenburg综合征患者基因突变分析[J].中华医学遗传学杂志, 2016, 33(3):312-315. doi: 10.3760/cma.j.issn.1003-9406.2016.03.007
[13] 刘亚兰, 张华, 冯永.神经嵴发育异常导致综合征型耳聋的机制[J].遗传, 2014, 36(11):1131-1144. https://www.cnki.com.cn/Article/CJFDTOTAL-YCZZ201411007.htm
[14] Lang D, Epstein JA.Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer[J]. Hum Mol Genet, 2003, 12(8):937-945. doi: 10.1093/hmg/ddg107
[15] Jalilian N, Tabatabaiefar MA, Farhadi M, et al.A novel mutation in the PAX3 gene causes Waardenburg syndrome type I in an Iranian family[J]. Int J Pediatr Otorhinolaryngol, 2015, 79(10):1736-1740. doi: 10.1016/j.ijporl.2015.07.039
[16] Song J, Feng Y, Acke FR, et al.Hearing loss in Waardenburg syndrome:a systematic review[J]. Clin Genet, 2016, 89(4):416-425. doi: 10.1111/cge.12631
[17] Li W, Mei L, Chen H, et al.New Genotypes and Phenotypes in Patients with 3 Subtypes of Waardenburg Syndrome Identified by Diagnostic Next-Generation Sequencing[J]. Neural Plast, 2019, 2019:7143458.
[18] Bocángel M, Melo US, Alves LU, et al.Waardenburg syndrome:Novel mutations in a large Brazilian sample[J]. Eur J Med Genet, 2018, 61(6):348-354. doi: 10.1016/j.ejmg.2018.01.012
[19] Merchant SN, McKenna MJ, Baldwin CT, et al.Otopathology in a case of type I Waardenburg's syndrome[J]. Ann Otol Rhinol Laryngol, 2001,110(9):875-882. doi: 10.1177/000348940111000913
[20] Shelby MV.Waardenburg Syndrome Expression and Penetrance[J]. J Rare Dis Res Treat, 2017, 2(6):31-40. doi: 10.29245/2572-9411/2017/6.1118
[21] Da CP, Menezes J, Romao L.The role of alternative splicing coupled to nonsense-mediated mRNA decay in human disease[J]. Int J Biochem Cell Biol, 2017, 91(Pt B):168-175.
-
 下载:
下载:


图(8)
计量
- 文章访问数: 1097
- PDF下载数: 500
- 施引文献: 0