
The value of genetic diagnosis of deafness in evaluating the prognosis of cochlear implantation
-
Abstract: Congenital deafness is known as the most common birth defect, and most sufferers from it manifest sensorineural hearing loss (SNHL), with hereditary factors responsible for approximately 60% of the cases of deafness. At present, cochlear implantation (CI) is regarded as the most mature and effective solution to treating severe and extremely severe SNHL. However, the outcome of implantation varies due to different genetic factors. With whole genome sequencing advancing, more deafness mutant genes and their types have been identified, which is conducive to clarifying the efficacy of CI in the patients with different mutations for clinical practice. This paper is aimed to summarize the different effects of CI on hereditary deafness and the potential mechanism discovered in recent years, and to clarify the role played by the genetic diagnosis of deafness in evaluating the efficacy of cochlear implantation.
-
Key words:
- congenital deafness /
- genetic diagnosis /
- cochlear implantation
-
-
表 1 POU3F4突变患者的人工耳蜗植入结果
术后效果 基因型(均为半合子) 发病年龄/岁 植入年龄/岁 评价标准 参考文献 好 c.383delG(p.G128fs) 3.0 15 开放式定词测验 Lee等[9] 好 X362TrpextX106 0.5 2.25 标准化语言测试 Kim等[14] 好 c.669T>A(p.Y223X) 0 5 语言学习 Su等[15] 好 c.950T>A(p.L317X) - - CAP、SIR、言语感知 Wu等[16] 欠佳 c.927-929del CTC(p.S310del) 2.5 3.7 闭合式单音节、开放式字识别 Stankovic等[10] 欠佳 c.C76>T(p.Q26*) 0 13 声音刺激、语言能力 Wu等[11] 差 623T>A(p.L208X) 1.25 6 智力、声音、言语、注意障碍 Lee等[9] 差 c.499c>T(p.R167X) 0.25 1.6 闭合式单音节,开放式字识别 Stankovic等[10] 差 c.623T>A,Xq21.2(80851535-82597832bp),Xq21.2(81810457-82810060bp),c.1060delA,c.1084T>C,c.910C>A,c.686A>G 0.28 4.8 CAP、智力 Choi等[12]  下载: 导出CSV
下载: 导出CSV
表 2 TMPRSS3突变患者的人工耳蜗植入结果
术后效果 基因型 发病年龄/岁 植入年龄/岁 评价标准 参考文献 好 c.207delC/c.916G>A,c.595G>A/ c.916G>A 语前 - 言语感知测试 Weegerink等[20] 好 c.325C>T/c.916G>A 语前 5 开放式句子测试 Chung等[23] 好 c.208delC纯合子 0 1.36 言语感知测试、主流学校 Battelino等[24] 好 c.916G>A/c.1250G>A 2 3 语言能力 Gao等[25] 好 c.325C>T/c.916G>A 语后 11 开放式句子测试 Chung等[23] 好 c.413C>A/c.916G>A,c.413C>A/c.323-6G>A,c.207delC/c.1276G>A,c.413C>A/c.595G>A 语后 - 言语感知测试 Weegerink等[20] 好 c.646C>T/c.916G>A 语后 6~20 - Elbracht等[26] 好 c.323-6G>A/c.916G>A 9 14 语言能力 Gao等[25] 好 c.390C>G/c.647G>T,c.226C>T/c.778G>A,c.212T>C/617-3-4dupAT 23 47.7 日语单音节测试 Miyagawa等[27] 好 c.36delC/c.916G>A 3 6 开放式句子测试 Gao等[28] 差 c.607C>T/c.778G>A,c.647G>T/c.771G>T - 42 听力保留评分 Yoshimura等[29] 差 c.413C>A/c.646C>T,c.413C>A/c.916G>A 5.5 39.5 辅音-核心辅音测试 Eppsteiner等[21] 差 c.208delC/c.1273G>A,c.413C>A/c.1273G>A,c.1345-2A>G纯合子 语后 51.7 ECochG Shearer等[22]
下载: 导出CSV
表 4 KNCQ1突变患者的人工耳蜗植入结果
术后效果 基因型 发病年龄/岁 植入年龄/岁 评价标准 参考文献 好 c.1686-1G>A纯合子 - 3.25 听力康复 Chorbachi等[38] 好 QVLQT1纯合子 1.4 3.0 开放式词组识别、词组理解、词汇能力 Berrettini等[39] 好 p.R518X/c.572delTGCGC,p.Q530X纯合子,c.572delTGCGC纯合子 0 1.5 听觉感知、开放式测试、婴幼儿言语听觉反应评估 Siem等[37] 好 c.DelGCGCC/c.1134delC - 49.0 ECochG Shearer等[22] 好 c.1741A>T/c.477+5G>A 0 2.7 日常生活听觉 Qiu等[40] 欠佳 c.572delTGCGC纯合子 3 3.1 句中主要词 Siem等[37]
下载: 导出CSV
表 5 USH1突变患者的人工耳蜗植入结果
术后效果 基因型 发病年龄/岁 植入年龄/岁 评价标准 参考文献 好 MYO7A:p.R1240Q纯合子,p.R1602Q&1170K/ p.R1240Q - 8.3 G(C)BI、EHL(等效听力水平) Pennings等[44] 好 MYO7A:c.4477G>A杂合子,c.0652G>A杂合子 - 19.0 听力保留评分 Yoshimura等[29] 欠佳 MYO7A:p.R666X/p.R302H,p.R1602Q&1170K/p.R212H - 21.7 G(C)BI、EHL Pennings等[44] 好 CDH23:p.P240L/p.R301Q,p.D1216A&V1807M/p.Q1716P 0.3 4.0 单词识别、婴幼儿言语听觉反应评估 Usami等[45] 好 CDH23:c.4762C>T纯合子,c.719C>T/c.902C>T,c.719C>T/c.6085C>T,c.2866C>T/c.4762C>T,c.719C>T/c.2866C>T - 34.0 听力保留评分 Yoshimura等[29] 欠佳 CDH23:p.R1305X纯合子 语前 6.0 IT-MAIS(婴幼儿听力整合问卷)、CAP、QACIU(CI使用评估) Liu等[43] 差 CDH23:p.R1305X纯合子 语前 7.0 IT-MAIS、CAP、QACIU Liu等[43] 差 CDH23:IVS20+1G>A杂合子 - 20.1 G(C)BI、EHL Pennings等[44] 差 PCDH15:c.145G>T杂合子,c.4744delC杂合子,c.1863-1864dup纯合子,c.4320-4328dup/c.3451G>A,c.4812G>T纯合子 - 2.9 CAP、SIR、言语感知 Wu等[16] 欠佳 PCDH15/CDH23:16delT/c.9565C>T 语前 11.0 IT-MAIS、CAP、QACIU Liu等[43]
下载: 导出CSV
-
[1] 苏钰, 戴朴. 耳聋基因诊断在人工耳蜗植入中的应用[J]. 中华耳科学杂志, 2018, 16(6): 785-790. doi: 10.3969/j.issn.1672-2922.2018.06.009
[2] Liu W, Edin F, Blom H, et al. Super-resolution structured illumination fluorescence microscopy of the lateral wall of the cochlea: the Connexin26/30 proteins are separately expressed in man[J]. Cell Tissue Res, 2016, 365(1): 13-27. doi: 10.1007/s00441-016-2359-0
[3] Cohen-Salmon M, Ott T, Michel V, et al. Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death[J]. Curr Biol, 2002, 12(13): 1106-1111. doi: 10.1016/S0960-9822(02)00904-1
[4] Chen S, Sun Y, Lin X, et al. Down regulated connexin26 at different postnatal stage displayed different types of cellular degeneration and formation of organ of Corti[J]. Biochem Biophys Res Commun, 2014, 445(1): 71-77. doi: 10.1016/j.bbrc.2014.01.154
[5] Roux I, Safieddine S, Nouvian R, et al. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse[J]. Cell, 2006, 127(2): 277-289. doi: 10.1016/j.cell.2006.08.040
[6] Rouillon I, Marcolla A, Roux I, et al. Results of cochlear implantation in two children with mutations in the OTOF gene[J]. Int J Pediatr Otorhinolaryngol, 2006, 70(4): 689-696. doi: 10.1016/j.ijporl.2005.09.006
[7] Nishio SY, Takumi Y, Usami SI. Laser-capture micro dissection combined with next-generation sequencing analysis of cell type-specific deafness gene expression in the mouse cochlea[J]. Hear Res, 2017, 348: 87-97. doi: 10.1016/j.heares.2017.02.017
[8] Phippard D, Lu L, Lee D, et al. Targeted mutagenesis of the POU-domain gene Brn4/Pou3f4 causes developmental defects in the inner ear[J]. J Neurosci, 1999, 19(14): 5980-5989. doi: 10.1523/JNEUROSCI.19-14-05980.1999
[9] Lee HK, Lee SH, Lee KY, et al. Novel POU3F4 mutations and clinical features of DFN3 patients with cochlear implants[J]. Clin Genet, 2009, 75(6): 572-575. doi: 10.1111/j.1399-0004.2009.01181.x
[10] Stankovic KM, Hennessey AM, Herrmann B, et al. Cochlear implantation in children with congenital X-linked deafness due to novel mutations in POU3F4 gene[J]. Ann Otol Rhinol Laryngol, 2010, 119(12): 815-822. doi: 10.1177/000348941011901205
[11] Wu HM, Jie HQ, Wang H, et al. A novel POU domain class 3 transcription factor 4 mutation causes X-linked non-syndromic hearing loss in a Chinese family[J]. Chin Med J(Engl), 2019, 132(18): 2251-2253.
[12] Choi BY, An YH, Song JJ, et al. Clinical observations and molecular variables of patients with hearing loss and incomplete partition type Ⅲ[J]. Laryngoscope, 2016, 126(3): E123-128. doi: 10.1002/lary.25573
[13] Choi BY, Kim DH, Chung T, et al. Destabilization and mislocalization of POU3F4 by C-terminal frameshift truncation and extension mutation[J]. Hum Mutat, 2013, 34(2): 309-316. doi: 10.1002/humu.22232
[14] Kim L, Wisely CE, Lucius S, et al. Positive Outcomes and Surgical Strategies for Bilateral Cochlear Implantation in a Child With X-Linked Deafness[J]. Ann Otol Rhinol Laryngol, 2016, 125(2): 173-176. doi: 10.1177/0003489415604167
[15] Su Y, Gao X, Huang SS, et al. Clinical and molecular characterization of POU3F4 mutations in multiple DFNX2 Chinese families[J]. BMC Med Genet, 2018, 19(1): 157. doi: 10.1186/s12881-018-0630-9
[16] Wu CC, Lin YH, Liu TC, et al. Identifying Children With Poor Cochlear Implantation Outcomes Using Massively Parallel Sequencing[J]. Medicine(Baltimore), 2015, 94(27): e1073.
[17] Kim HM, Wangemann P. Epithelial cell stretching and luminal acidification lead to a retarded development of stria vascularis and deafness in mice lacking pendrin[J]. PLoS One, 2011, 6(3): e17949. doi: 10.1371/journal.pone.0017949
[18] Guipponi M, Antonarakis SE, Scott HS. TMPRSS3, a type Ⅱ transmembrane serine protease mutated in non-syndromic autosomal recessive deafness[J]. Front Biosci, 2008, 13: 1557-1567. doi: 10.2741/2780
[19] Peng A, Li Y, Pan X, et al. Survival of Cochlear Spiral Ganglion Neurons Improved In vivo by Anti-miR204 via TMPRSS3 [J]. West Indian Med J, 2015, 65(2): 379-382.
[20] Weegerink NJ, Schraders M, Oostrik J, et al. Genotype-phenotype correlation in DFNB8/10 families with TMPRSS3 mutations[J]. J Assoc Res Otolaryngol, 2011, 12(6): 753-766. doi: 10.1007/s10162-011-0282-3
[21] Eppsteiner RW, Shearer AE, Hildebrand MS, et al. Prediction of cochlear implant performance by genetic mutation: the spiral ganglion hypothesis[J]. Hear Res, 2012, 292(1/2): 51-58.
[22] Shearer AE, Tejani VD, Brown CJ, et al. In Vivo Electrocochleography in Hybrid Cochlear Implant Users Implicates TMPRSS3 in Spiral Ganglion Function[J]. Sci Rep, 2018, 8(1): 14165. doi: 10.1038/s41598-018-32630-9
[23] Chung J, Park SM, Chang SO, et al. A novel mutation of TMPRSS3 related to milder auditory phenotype in Korean postlingual deafness: a possible future implication for a personalized auditory rehabilitation[J]. J Mol Med(Berl), 2014, 92(6): 651-663.
[24] Battelino S, Klancar G, Kovac J, et al. TMPRSS3 mutations in autosomal recessive nonsyndromic hearing loss[J]. Eur Arch Otorhinolaryngol, 2016, 273(5): 1151-1154. doi: 10.1007/s00405-015-3671-0
[25] Gao X, Huang SS, Yuan YY, et al. Identification of TMPRSS3 as a Significant Contributor to Autosomal Recessive Hearing Loss in the Chinese Population[J]. Neural Plast, 2017, 2017: 3192090.
[26] Elbracht M, Senderek J, Eggermann T, et al. Autosomal recessive postlingual hearing loss(DFNB8): compound heterozygosity for two novel TMPRSS3 mutations in German siblings[J]. J Med Genet, 2007, 44(6): e81. doi: 10.1136/jmg.2007.049122
[27] Miyagawa M, Nishio SY, Sakurai Y, et al. The patients associated with TMPRSS3 mutations are good candidates for electric acoustic stimulation[J]. Ann Otol Rhinol Laryngol, 2015, 124 Suppl 1: 193S-204S.
[28] Gao X, Yuan YY, Wang GJ, et al. Novel Mutations and Mutation Combinations of TMPRSS3 Cause Various Phenotypes in One Chinese Family with Autosomal Recessive Hearing Impairment[J]. Biomed Res Int, 2017, 2017: 4707315.
[29] Yoshimura H, Moteki H, Nishio SY, et al. Genetic testing has the potential to impact hearing preservation following cochlear implantation[J]. Acta Otolaryngol, 2020, 140(6): 438-444. doi: 10.1080/00016489.2020.1730439
[30] Delmaghani S, del Castillo FJ, Michel V, et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy[J]. Nat Genet, 2006, 38(7): 770-778. doi: 10.1038/ng1829
[31] Cryns K, Thys S, Van Laer L, et al. The WFS1 gene, responsible for low frequency sensorineural hearing loss and Wolfram syndrome, is expressed in a variety of inner ear cells[J]. Histochem Cell Biol, 2003, 119(3): 247-256. doi: 10.1007/s00418-003-0495-6
[32] Rigoli L, Lombardo F, Di Bella C. Wolfram syndrome and WFS1 gene[J]. Clin Genet, 2011, 79(2): 103-117. doi: 10.1111/j.1399-0004.2010.01522.x
[33] Liu WH, Chang PY, Chang SC, et al. Mutation screening in non-syndromic hearing loss patients with cochlear implantation by massive parallel sequencing in Taiwan[J]. PLoS One, 2019, 14(1): e0211261. doi: 10.1371/journal.pone.0211261
[34] Hogewind BF, Pennings RJ, Hol FA, et al. Autosomal dominant optic neuropathy and sensorineual hearing loss associated with a novel mutation of WFS1[J]. Mol Vis, 2010, 16: 26-35.
[35] Häkli S, Kytövuori L, Luotonen M, et al. WFS1 mutations in hearing-impaired children[J]. Int J Audiol, 2014, 53(7): 446-451. doi: 10.3109/14992027.2014.887230
[36] Wangemann P, Liu J, Marcus DC. Ion transport mechanisms responsible for K+ secretion and the transepithelial voltage across marginal cells of stria vascularis in vitro[J]. Hear Res, 1995, 84(1/2): 19-29.
[37] Siem G, Früh A, Leren TP, et al. Jervell and Lange-Nielsen syndrome in Norwegian children: aspects around cochlear implantation, hearing, and balance[J]. Ear Hear, 2008, 29(2): 261-269. doi: 10.1097/AUD.0b013e3181645393
[38] Chorbachi R, Graham JM, Ford J, et al. Cochlear implantation in Jervell and Lange-Nielsen syndrome[J]. Int J Pediatr Otorhinolaryngol, 2002, 66(3): 213-221. doi: 10.1016/S0165-5876(02)00181-7
[39] Berrettini S, Forli F, Ursino F, et al. Cochlear implant in Jervell and Lange-Nielsen syndrome[J]. Audiological Medicine, 2003, 1(4): 224-227. doi: 10.1080/16513860310001924
[40] Qiu Y, Chen S, Wu X, et al. Jervell and Lange-Nielsen Syndrome due to a Novel Compound Heterozygous KCNQ1 Mutation in a Chinese Family[J]. Neural Plast, 2020, 2020: 3569359.
[41] Zallocchi M, Meehan DT, Delimont D, et al. Role for a novel Usher protein complex in hair cell synaptic maturation[J]. PLoS One, 2012, 7(2): e30573. doi: 10.1371/journal.pone.0030573
[42] Cosgrove D, Zallocchi M. Usher protein functions in hair cells and photoreceptors[J]. Int J Biochem Cell Biol, 2014, 46: 80-89. doi: 10.1016/j.biocel.2013.11.001
[43] Liu XZ, Angeli SI, Rajput K, et al. Cochlear implantation in individuals with Usher type 1 syndrome[J]. Int J Pediatr Otorhinolaryngol, 2008, 72(6): 841-847. doi: 10.1016/j.ijporl.2008.02.013
[44] Pennings RJ, Damen GW, Snik AF, et al. Audiologic performance and benefit of cochlear implantation in Usher syndrome type Ⅰ[J]. Laryngoscope, 2006, 116(5): 717-722. doi: 10.1097/01.mlg.0000205167.08415.9e
[45] Usami S, Miyagawa M, Nishio SY, et al. Patients with CDH23 mutations and the 1555A>G mitochondrial mutation are good candidates for electric acoustic stimulation(EAS)[J]. Acta Otolaryngol, 2012, 132(4): 377-384. doi: 10.3109/00016489.2011.649493
[46] Brookes JT, Kanis AB, Tan LY, et al. Cochlear implantation in deafness-dystonia-optic neuronopathy(DDON)syndrome[J]. Int J Pediatr Otorhinolaryngol, 2008, 72(1): 121-126. doi: 10.1016/j.ijporl.2007.08.019
[47] Roesch K, Hynds PJ, Varga R, et al. The calcium-binding aspartate/glutamate carriers, citrin and aralar1, are new substrates for the DDP1/TIMM8a-TIMM13 complex[J]. Hum Mol Genet, 2004, 13(18): 2101-2111. doi: 10.1093/hmg/ddh217
[48] Cif L, Gonzalez V, Garcia-Ptacek S, et al. Progressive dystonia in Mohr-Tranebjaerg syndrome with cochlear implant and deep brain stimulation[J]. Mov Disord, 2013, 28(6): 737-738. doi: 10.1002/mds.25519
[49] Egilmez OK, Kalcioglu MT. Genetics of Nonsyndromic Congenital Hearing Loss[J]. Scientifica(Cairo), 2016, 2016: 7576064.
[50] 黄玉宇, 程岚, 杨军, 等. 耳蜗神经发育不良患儿人工耳蜗植入术前影像和电生理评估[J]. 临床耳鼻咽喉头颈外科杂志, 2019, 33(8): 729-735. https://www.cnki.com.cn/Article/CJFDTOTAL-LCEH201908012.htm
[51] Miya SJ, Georgie C, Amanda B, et al. Predicting speech-sound disorder outcomes in school-age children with hearing loss: The VicCHILD experience[J]. Int J Lang Commun Disord, 2020, 55(4): 537-546. doi: 10.1111/1460-6984.12536
-
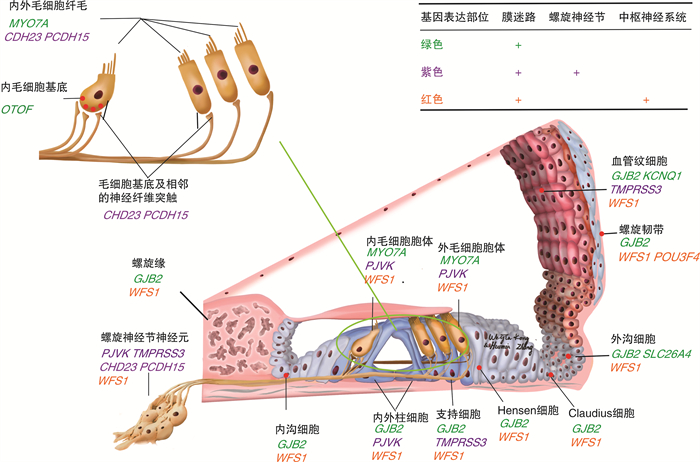
图(1)
表(6)
计量
- 文章访问数: 1872
- PDF下载数: 721
- 施引文献: 0