
Identification of a novel mutation of CEACAM16 gene in a Chinese family with autosomal dominant nonsyndromic hearing loss
-
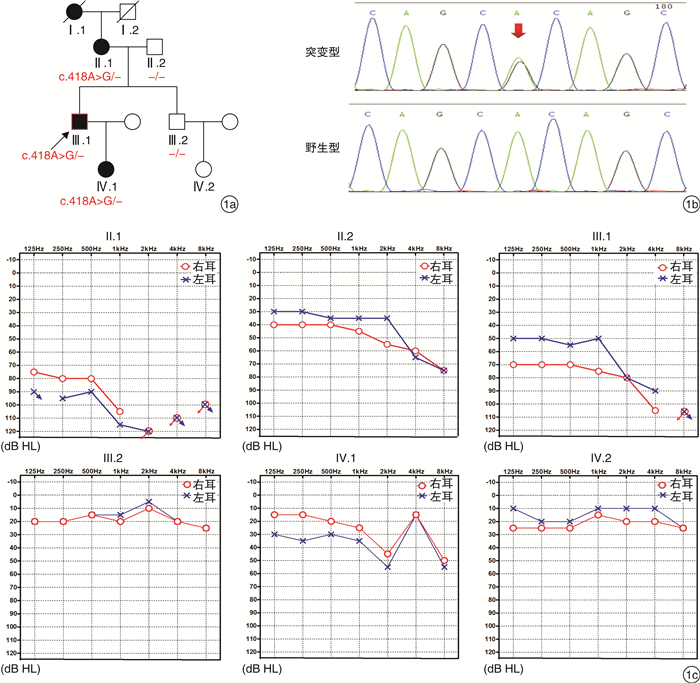
摘要: 目的 对我国一个常染色体显性遗传性耳聋家系进行临床特征及分子遗传学分析,明确其病因,为今后临床上开展基因诊断奠定基础。方法 通过问卷调查方式采集家系成员的病史,并进行系统的查体和听力学检查。家系成员留取外周静脉血提取DNA,开展包括139个已知耳聋基因的目标区域捕获和测序,同时对6个耳聋相关的线粒体DNA突变位点进行检测。结果 该家系成员主要表现为20岁左右发病的听力进行性下降的常染色体显性遗传性耳聋,听力图主要表现为高频下降型。基因检测结果提示先证者及其母亲和女儿携带CEACAM16基因c.418A>G /p.Thr140Ala杂合突变,并且其基因型与临床表型共分离,考虑为新发现的致病突变。结论 本研究在我国的一个常染色体显性遗传性耳聋家系中发现了CEACAM16基因的一个新的突变位点,这是我国耳聋人群中检测到的第2个可能的致病突变,丰富了我国人群该基因的突变图谱。
-
关键词:
- CEACAM16基因 /
- 耳聋 /
- 基因突变
Abstract: Objective To explore the genetic cause of a Chinese autosomal dominant nonsyndromic hearing loss family and investigate the clinical features and molecular genetic characteristics of this family.Method Detailed medical history and systematic audiology tests were carried out in the family members, and they were subjected to comprehensive genetic analyses using massively parallel sequencing, which targeted 139 known deafness genes and 6 mitochondrial DNA mutations associated with hearing loss.Result This family's hearing loss was consistent with autosomal dominant nonsyndromic hearing loss. The affected family members appeared to have developed a high-frequency hearing loss with the onset of twenties. We identified a heterozygous missense mutation, c.418A>G/p. Thr140Ala in the CEACAM16 gene, segregating with the deafness in this family.Conclusion In this study, we identified a new mutation of CEACAM16 gene, which was the second mutation identified in Chinese hearing loss population. It has enriched the mutation spectrum of this gene.-
Key words:
- CEACAM16 gene /
- deafness /
- mutation
-
-
表 1 CEACAM16基因已知致病突变及其临床表型汇总
外显子 碱基改变 氨基酸改变 国家 发病年龄 家系表型 参考文献 Exon2 c.37G>T 伊朗 12~13岁 迟发性进行性听力下降的常染色体隐性遗传性耳聋家系 Booth 〔8〕 Exon4 c.418A>C p.Thr140Pro 美国 10余岁 迟发性进行性听力下降的常染色体显性遗传性耳聋家系 Zheng〔2〕 Exon4 c.418A>G p.Thr140Ala 中国 20~24岁 迟发性进行性听力下降的常染色体显性遗传性耳聋家系 本研究 Exon4 c.436C>T p.Arg146Ter 巴西 10余岁 迟发性进行性听力下降的常染色体隐性遗传性耳聋家系 Dias〔9〕 Exon4 c.505G>A p.Gly169Arg 中国 10~25岁 迟发性进行性听力下降的常染色体显性遗传性耳聋家系 Wang〔3〕 Exon6 c.1094T>G p.Leu365Arg 德国 11岁 散发患者,de novo突变 Hofrichter〔7〕 c.662-1G>C 伊朗 10~13岁 迟发性进行性听力下降的常染色体隐性遗传性耳聋家系 Booth〔8〕  下载: 导出CSV
下载: 导出CSV
-
[1] 王秋菊, 刘穹. 常染色体显性遗传性耳聋倡(1)[J]. 听力学及言语疾病杂志, 2016, 24(2): 213-216. doi: 10.3969/j.issn.1006-7299.2016.02.027
[2] Zheng J, Miller KK, Yang T, et al. Carcinoembryonic antigen-related cell adhesion molecule 16 interacts with alpha-tectorin and is mutated in autosomal dominant hearing loss(DFNA4)[J]. Proc Natl Acad Sci U S A, 2011, 108(10): 4218-4223. doi: 10.1073/pnas.1005842108
[3] Wang H, Wang X, He C, et al. Exome sequencing identifies a novel CEACAM16 mutation associated with autosomal dominant nonsyndromic hearing loss DFNA4B in a Chinese family[J]. J Hum Genet, 2015, 60(3): 119-126. doi: 10.1038/jhg.2014.114
[4] 王欣炜. CEACAM16基因新突变导致遗传性耳聋的分子机制研究[C]. 湖南: 中南大学, 2014.
[5] Kammerer R, Rüttiger L, Riesenberg R, et al. Loss of mammal-specific tectorial membrane component carcinoembryonic antigen cell adhesion molecule 16(CEACAM16) leads to hearing impairment at low and high frequencies[J]. J Biol Chem, 2012, 287(26): 21584-21598. doi: 10.1074/jbc.M111.320481
[6] Chen AH, Ni L, Fukushima K, et al. Linkage of a gene for dominant non-syndromic deafness to chromosome 19[J]. Hum Mol Genet, 1995, 4(6): 1073-1076. doi: 10.1093/hmg/4.6.1073
[7] Hofrichter MA, Nanda I, Gräf J, et al. A Novel de novo Mutation in CEACAM16 Associated with Postlingual Hearing Impairment[J]. Mol Syndromol, 2015, 6(4): 156-163. doi: 10.1159/000439576
[8] Booth KT, Kahrizi K, Najmabadi H, et al. Old gene, new phenotype: splice-altering variants in CEACAM16 cause recessive non-syndromic hearing impairment[J]. J Med Genet, 2018, 55(8): 555-560. doi: 10.1136/jmedgenet-2018-105349
[9] Dias A, Lezirovitz K, Nicastro FS, et al. Further evidence for loss-of-function mutations in the CEACAM16 gene causing nonsyndromic autosomal recessive hearing loss in humans[J]. J Hum Genet, 2019, 64(3): 257-260. doi: 10.1038/s10038-018-0546-4
-

图(2)
表(1)
计量
- 文章访问数: 948
- PDF下载数: 1004
- 施引文献: 0