
A case report of congenital sensorineural deafness caused by novel mutation in Usher1C and related literature analysis
-
-
关键词:
- 听觉丧失,感音神经性 /
- Usher1C /
- 常染色体隐性遗传 /
- DFNB18
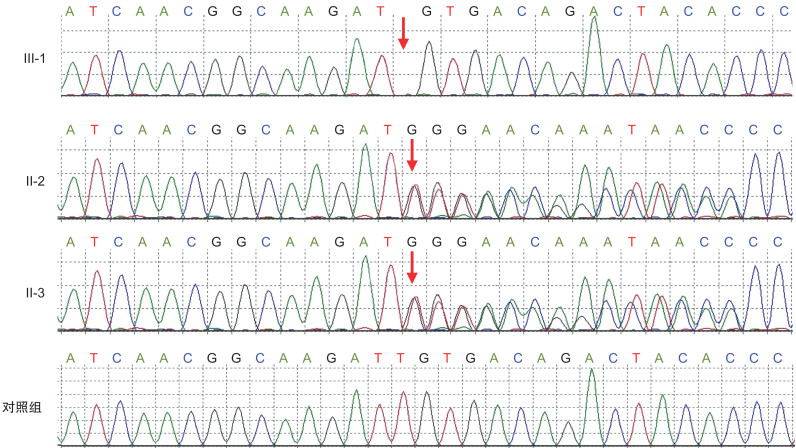
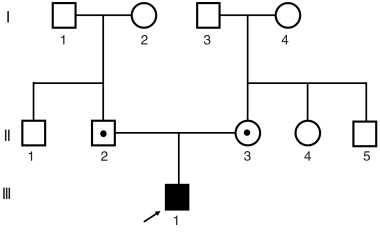
Abstract: To study the clinical features and causes of congenital Usher hearing loss in one child. Clinical examination, audiological tests, visual acuity examination were conducted in the proband and its family members, and second-generation sequencing technology for deafness gene detection was employed. The proband exhibited profound sensorineural deafness(hearing threshold>90 dB nHL). There was no visual loss after follow-up. Other family members had no history of hearing loss. The gene test indicated that the proband had a frameshift mutation for the thymine(T) deletion at the 1527 site of the Usher1C gene. The mutation was a homozygous mutation, and was from the father and the mother, respectively, which caused the truncation of the encoded protein. Normal function, Usher syndrome or non-syndromic deafness DFNB18 can occur. This is the first case in China demonstrating congenital deafness due to homozygous mutation of Usher1C gene c. 1527delT. This study enriches the gene spectrum of deafness in China.-
Key words:
- hearing loss, sensorineural /
- Usher1C /
- autosomal recessive inheritance /
- DFNB18
-
-
[1] 袁永一, 戴朴. 遗传性聋的精准医疗[J]. 临床耳鼻咽喉头颈外科杂志, 2016, 30(1): 1-5. https://www.cnki.com.cn/Article/CJFDTOTAL-LCEH201601001.htm
[2] Jansen F, Kalbe B, Scholz P, et al. Impact of the Usher syndrome on olfaction[J]. Hum Mol Genet, 2016, 25(3): 524-533. doi: 10.1093/hmg/ddv490
[3] Zhao Y, Hosono K, Suto K, et al. The first USH2A mutation analysis of Japanese autosomal recessiveretinitis pigmentosa patients: a totally different mutation profile with the lack of frequent mutations found in Caucasian patients[J]. J Hum Genet, 2014, 59(9): 521-528. doi: 10.1038/jhg.2014.65
[4] Sorusch N, Bau K, Plutniok J, et al. Characterization of the ternary Usher syndrome SANS/ush2a/whirlinprotein complex[J]. Hum Mol Genet, 2017, 26(6): 1157-1172.
[5] Zou J, Chen Q, Almishaal A, et al. The roles of USH1 proteins and PDZ domain-containing USH proteins in USH2 complex integrity in cochlear hair cells[J]. Hum Mol Genet, 2017, 26(3): 624-636.
[6] Magliulo G, Iannella G, Gagliardi S, et al. Usher's Syndrome Type Ⅱ: Usher's Syndrome Type Ⅱ: A Comparative Study of Genetic Mutations and Vestibular System Evaluation[J]. Otolaryngol Head Neck Surg, 2017, 157(5): 853-860. doi: 10.1177/0194599817715235
[7] Liquori A, Vaché C, Baux D, et al. Whole USH2A Gene Sequencing Identifies Several New Deep IntronicMutations[J]. Hum Mutat, 2016, 37(2): 184-193. doi: 10.1002/humu.22926
[8] Eandi CM, Dallorto L, Spinetta R, et al. Targeted next generation sequencing in Italian patients with Ushersyndrome: phenotype-genotype correlations[J]. Sci Rep, 2017, 7(1): 15681-15681. doi: 10.1038/s41598-017-16014-z
[9] Ouyang XM, Xia XJ, Verpy E, et al. Mutations in the alternatively spliced exons of USH1C cause non-syndromic recessive deafness[J]. Hum Genet, 2002, 111(1): 26-30. doi: 10.1007/s00439-002-0736-0
[10] Koparir A, Karatas OF, Atayoglu AT, et al. Whole-exome sequencing revealed two novel mutations in Usher syndrome[J]. Gene, 2015, 563(2): 215-218. doi: 10.1016/j.gene.2015.03.060
-
 下载:
下载:
图(2)
计量
- 文章访问数: 773
- PDF下载数: 337
- 施引文献: 0